
If you have Ehlers-Danlos Syndrome and you're dealing with burning feet, pins and needles that come and go, or a strange temperature sensitivity that no one can explain — you are not imagining it. For years, the neurological side of EDS was dismissed as “just part of hypermobility” or “joint pain referred to the nerves.” That explanation is crumbling. Researchers have now documented small fiber neuropathy in a majority of hypermobile EDS patients, and the mechanism is finally starting to make sense.
I'm Janet Ellis, and I want to be clear from the start — I'm not a doctor. I'm a patient advocate who lives with peripheral neuropathy and spends my days translating research into language that actually helps people. EDS is a connective tissue disorder, not a nerve disorder on its face. But connective tissue is in everything, including the protective sheaths around nerves. When the scaffolding that supports your nerves is fragile, the nerves themselves pay a price. This article walks through what we know, what the testing looks like, and what it changes about treatment.
The Connection Doctors Missed for Decades
Ehlers-Danlos Syndrome is a group of inherited connective tissue disorders — 13 recognized subtypes as of the 2017 International Classification. The most common by far is hypermobile EDS (hEDS), and alongside it sits hypermobility spectrum disorder (HSD), which has overlapping symptoms without meeting every hEDS criterion. Together, these two are where most of the neuropathy research is happening.
Up to 78% of hypermobile EDS patients have reduced intraepidermal nerve fiber density consistent with small fiber neuropathy — biopsy-confirmed nerve damage that most of them have never been told about.
For a long time, when EDS patients described burning feet, electric-shock sensations, or numbness, the explanation was usually “compression from your joints” or “referred pain.” Some of that is real — subluxing joints can pinch nerves. But it didn't explain the patients whose MRI was clean, whose joints were stable that day, and whose symptoms were spreading in a pattern that looked like a stocking-glove neuropathy.
Starting around 2015, a small group of researchers did what should have been done decades earlier — they performed skin punch biopsies on EDS patients with unexplained burning pain. They counted the tiny nerve endings in the epidermis under a microscope. The measure is called intraepidermal nerve fiber density, or IENFD, and it's the gold standard for diagnosing small fiber neuropathy. The results were striking.
In one 2022 study published in the Journal of Internal Medicine, roughly 78% of hEDS patients had reduced IENFD consistent with small fiber neuropathy. Let that number sit for a moment. Three out of four people with hypermobile EDS have objective, biopsy-confirmed nerve damage that most of them have never been told about.
What Is Small Fiber Neuropathy and Why It Fits EDS
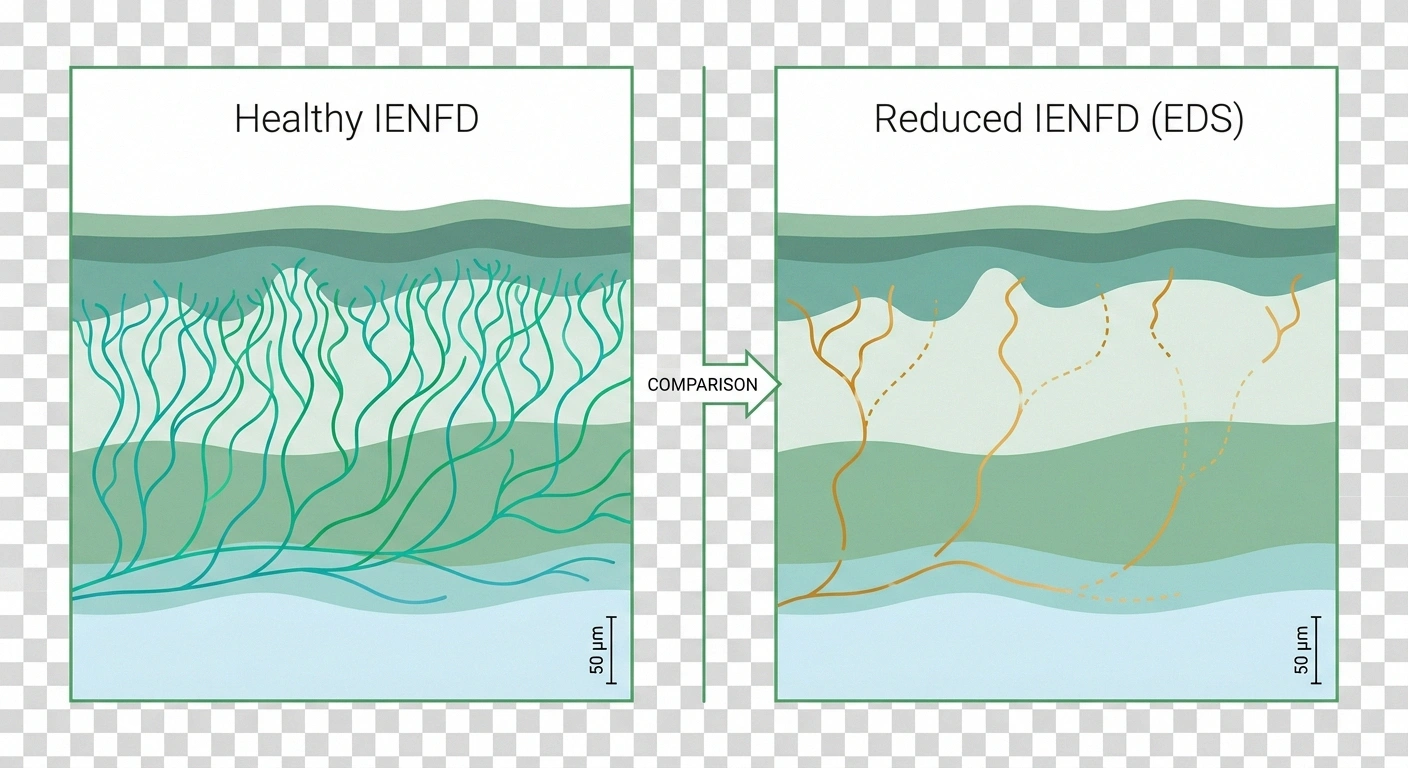

Peripheral nerves come in two broad categories. Large myelinated fibers carry vibration, proprioception (joint position sense), and motor signals — they're what a standard nerve conduction study measures. Small unmyelinated fibers carry temperature sensation, pain, light touch, and the autonomic signals that control sweating, blood flow, gut motility, and heart rate regulation.
of hypermobile EDS patients showed reduced intraepidermal nerve fiber density consistent with small fiber neuropathy
Fernandez et al., Journal of Internal Medicine, 2022
Small fiber neuropathy affects the second group. It's characterized by burning, tingling, pins and needles, and a strange mismatch with objective testing — because standard nerve conduction studies can come back completely normal while the person is genuinely suffering. The only way to see small fiber damage is skin biopsy or specialized autonomic testing.
Small fiber neuropathy fits the EDS picture for several reasons. The symptoms are diffuse rather than localized to a pinched spinal nerve. They often include autonomic features that overlap heavily with POTS (postural orthostatic tachycardia syndrome), which is itself common in EDS. And the pattern tends to be length-dependent at first — burning starts in the toes and fingertips — then can become generalized or patchy over time. None of that looks like a joint problem. All of it looks like neuropathy.
Why Connective Tissue Weakness Damages Nerves
The mechanism question — why does a collagen disorder cause nerve damage — doesn't have a single answer yet. Researchers have proposed several overlapping explanations, and they probably all contribute.
Why EDS Damages Nerves: Four Overlapping Mechanisms
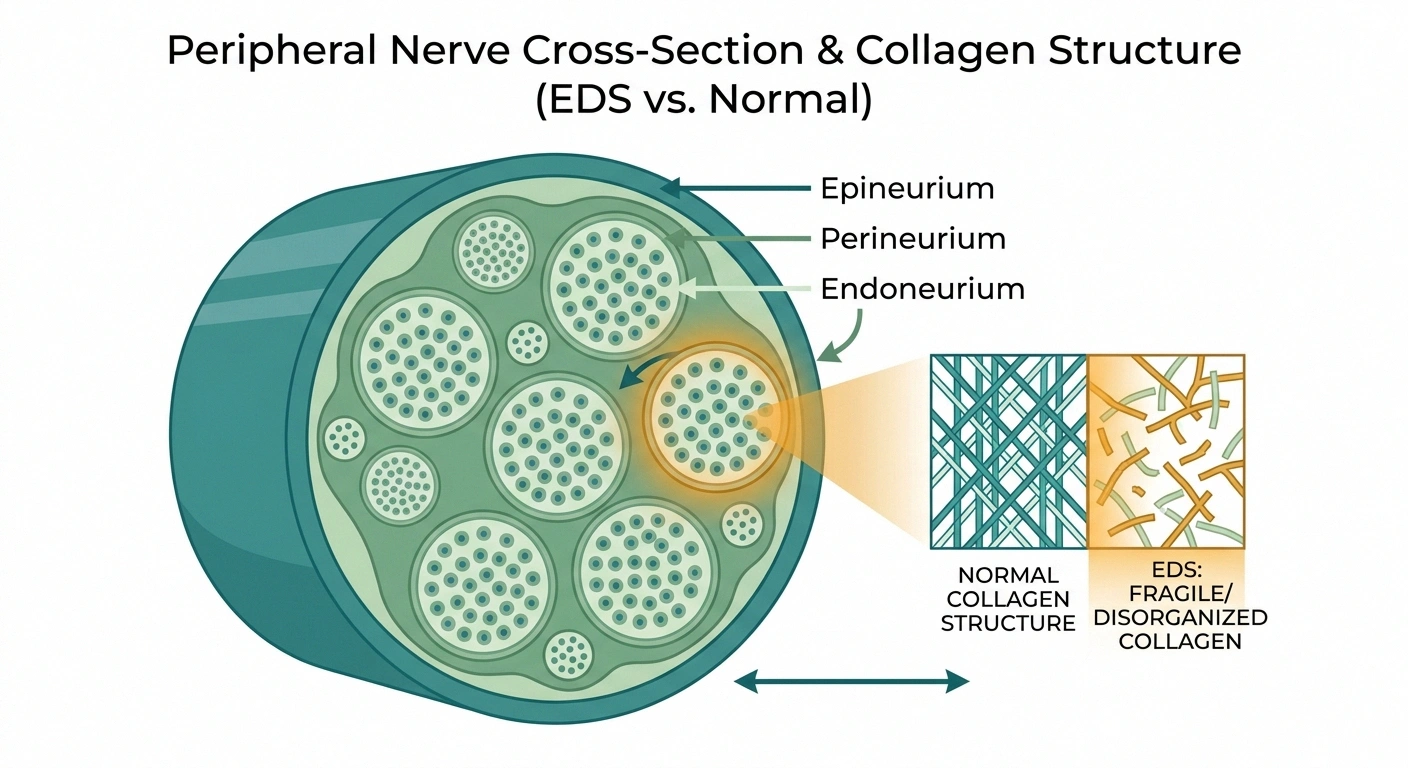
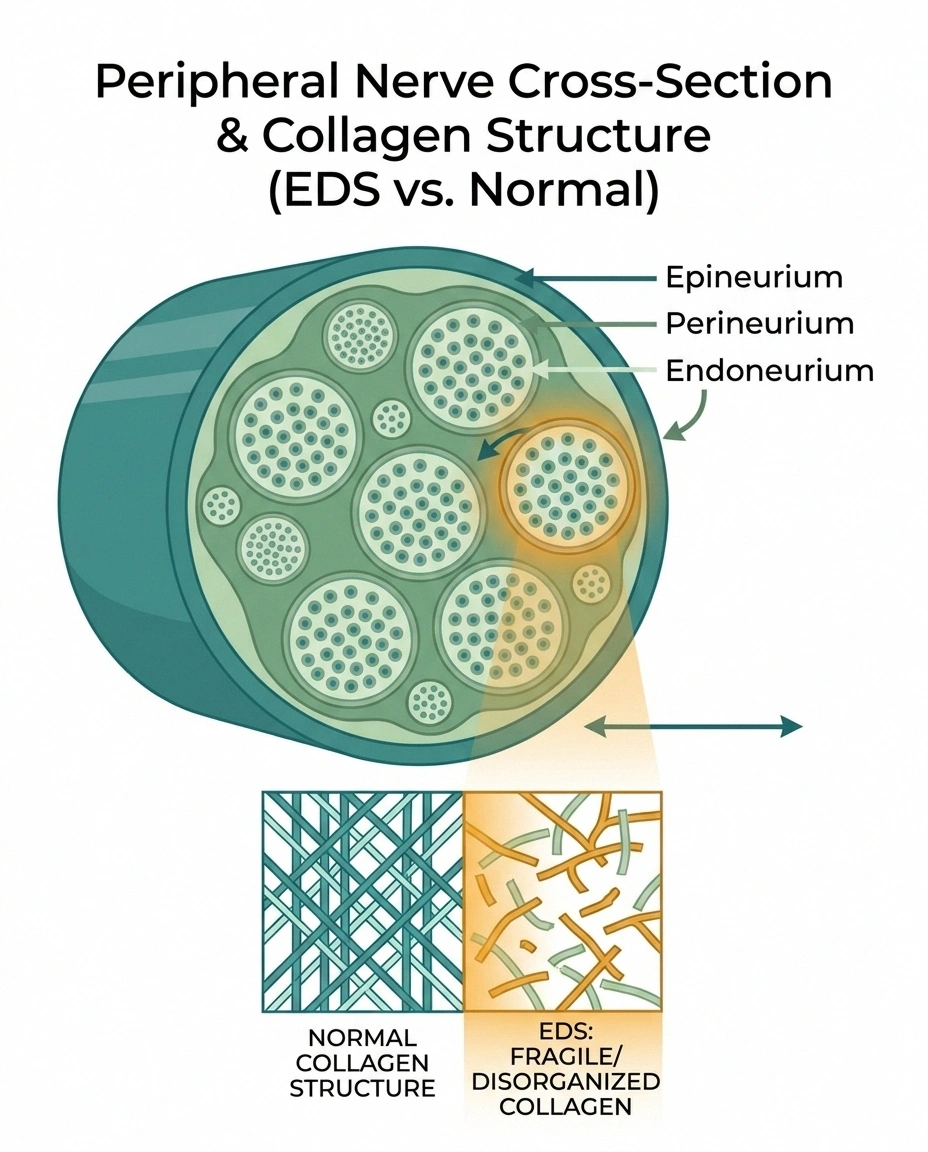
The first is structural. Peripheral nerves are wrapped in three layers of connective tissue — the endoneurium, perineurium, and epineurium. These sheaths are largely collagen. In EDS, collagen is abnormal. If the sheaths that protect your nerves are themselves fragile, minor mechanical stresses that a healthy nerve shrugs off can cause microdamage over years.
The second is vascular. Tiny blood vessels called the vasa nervorum supply oxygen and nutrients to peripheral nerves. In EDS, blood vessels tend to be fragile too. If the nerve's blood supply is chronically compromised, the smallest and most distal fibers — which are hardest to keep supplied — will fail first. That matches the distal-then-generalized pattern we see.
The third is mechanical. Hypermobile joints sublux, partially dislocate, and hyperextend repeatedly across a lifetime. Nerves that cross those joints are stretched, compressed, and irritated in ways they shouldn't be. Over time that repeated trauma shows up as nerve damage.
The fourth is autoimmune. A meaningful subset of small fiber neuropathy is driven by immune mechanisms. EDS patients have higher rates of several autoimmune and dysimmune conditions, including mast cell activation syndrome. An inflamed or dysregulated immune system attacking small fibers is a real possibility in a portion of these cases.
None of these mechanisms have to be singular. In a given patient, several may be acting at once, which is partly why the neuropathy in EDS can feel so bewildering — it doesn't behave exactly like a purely diabetic or purely toxic neuropathy.
The Symptoms Patients Describe
When EDS patients finally get a neurologist to take their nerve symptoms seriously, the descriptions tend to cluster in recognizable ways. These are the ones I hear most often from the community, and they line up with the published symptom clusters.
- ✓Burning feet or hands that feels like sunburn that won't cool down
- ✓Pins-and-needles or electric shocks, often worse at night
- ✓Bedsheets or sock seams that hurt (allodynia)
- ✓Ice-cold feet while the rest of you feels normal
- ✓Racing heart on standing, flushing, digestive slowness
Three or more of these alongside hypermobility is a signal worth a skin biopsy conversation with your neurologist.
Burning in the feet, the hands, or along patches of the skin. Some describe it as a sunburn that won't cool down. Others describe it as the feeling of having walked barefoot on hot pavement, except the pavement is gone.
Pins and needles, electric shocks, and prickling. These often come and go. They can be triggered by heat, cold, fatigue, menstruation, or nothing identifiable at all. They frequently get worse at night, which is a hallmark of small fiber involvement.
Hyperesthesia and allodynia. Hyperesthesia means ordinary touch feels magnified and uncomfortable. Allodynia means stimuli that shouldn't hurt — the weight of a bedsheet, the seam of a sock — actually do hurt. Both are classic small fiber neuropathy findings.
Temperature dysregulation. Hands and feet that run ice cold while the rest of the body is normal. Feet that feel hot when the room is cool. An inability to tell if bathwater is dangerously hot until the skin protests.
Autonomic symptoms. Orthostatic intolerance, racing heart on standing, digestive slowness, bladder urgency, skin flushing, sweat abnormalities. These aren't sensory, but they come from the same small unmyelinated fibers that carry burning pain signals. Where one shows up, the others often follow.
The Diagnostic Path

If you're an EDS patient and the symptoms above sound familiar, the workup has a specific shape. It starts with ruling out the common non-EDS causes of neuropathy, because the worst outcome is to blame EDS for damage that actually has a treatable cause you missed.
The EDS Small Fiber Neuropathy Workup
Basic blood work should look at blood sugar and hemoglobin A1c (diabetic neuropathy is the most common cause of neuropathy worldwide, EDS or not), vitamin B12 (low B12 is a correctable cause), thyroid function, kidney function, and a few basic autoimmune markers. A competent neurologist will order most of these without you having to ask.
Standard nerve conduction studies and EMG measure large fibers. These will often be normal in EDS-related small fiber neuropathy, and a normal result does not mean your nerves are fine. It means the test was looking at the wrong fibers.
The gold standard for small fiber neuropathy is a 3mm skin punch biopsy from the distal leg, processed at a laboratory that counts intraepidermal nerve fiber density per millimeter. Low density confirms small fiber neuropathy. Not every neurology clinic orders these, so you may need to ask directly or be referred to an academic center.
Autonomic testing is the third leg. Tilt-table testing, QSART (quantitative sudomotor axon reflex test), and COMPASS-31 (a validated symptom questionnaire) can identify autonomic small fiber involvement. If you have POTS-like symptoms alongside the burning and tingling, this is worth pursuing.
Why EDS Patients Need Drug Choices Made Carefully
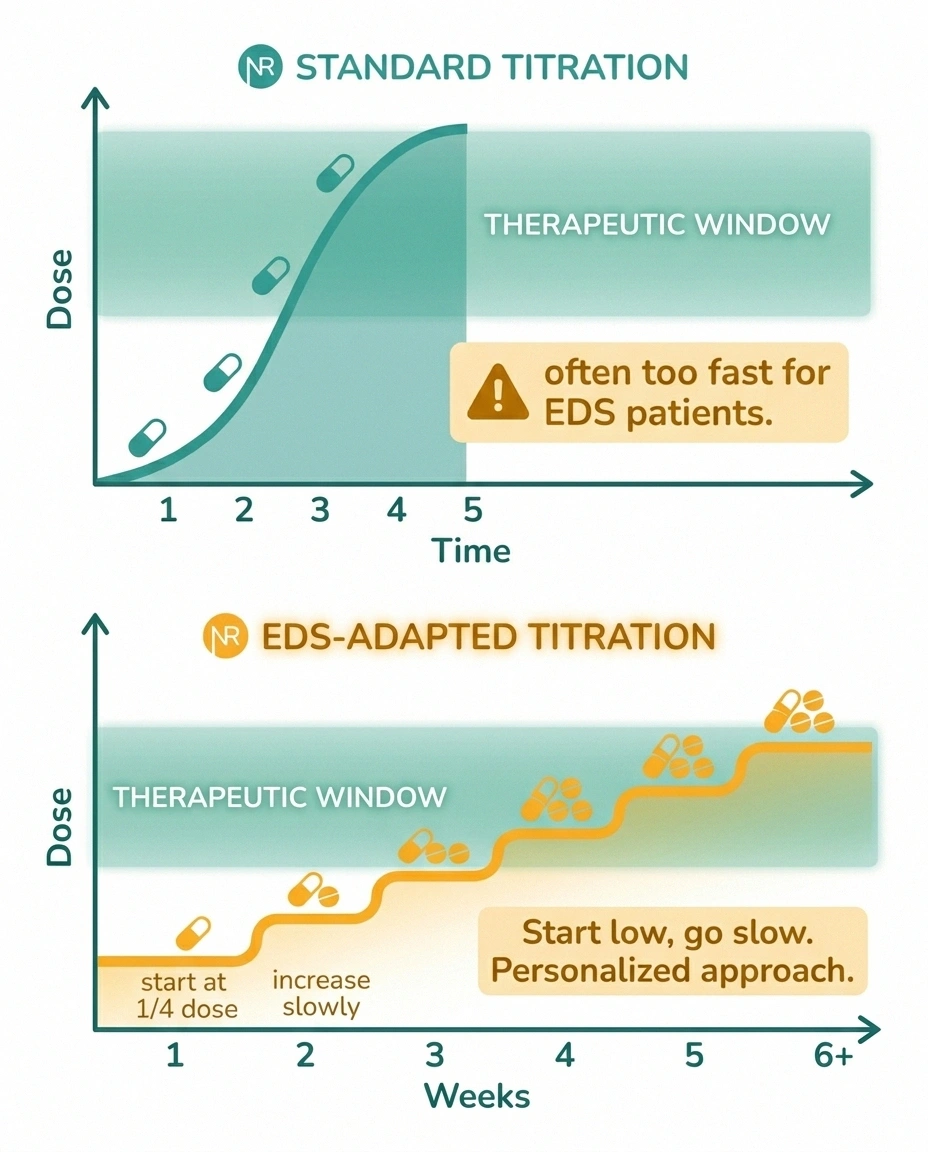
Once small fiber neuropathy is diagnosed, the treatment menu looks similar to neuropathy treatment in the general population, with some important modifications. EDS patients are often drug-sensitive, metabolize certain medications atypically, and have comorbidities (POTS, MCAS, GI dysmotility) that narrow the options.
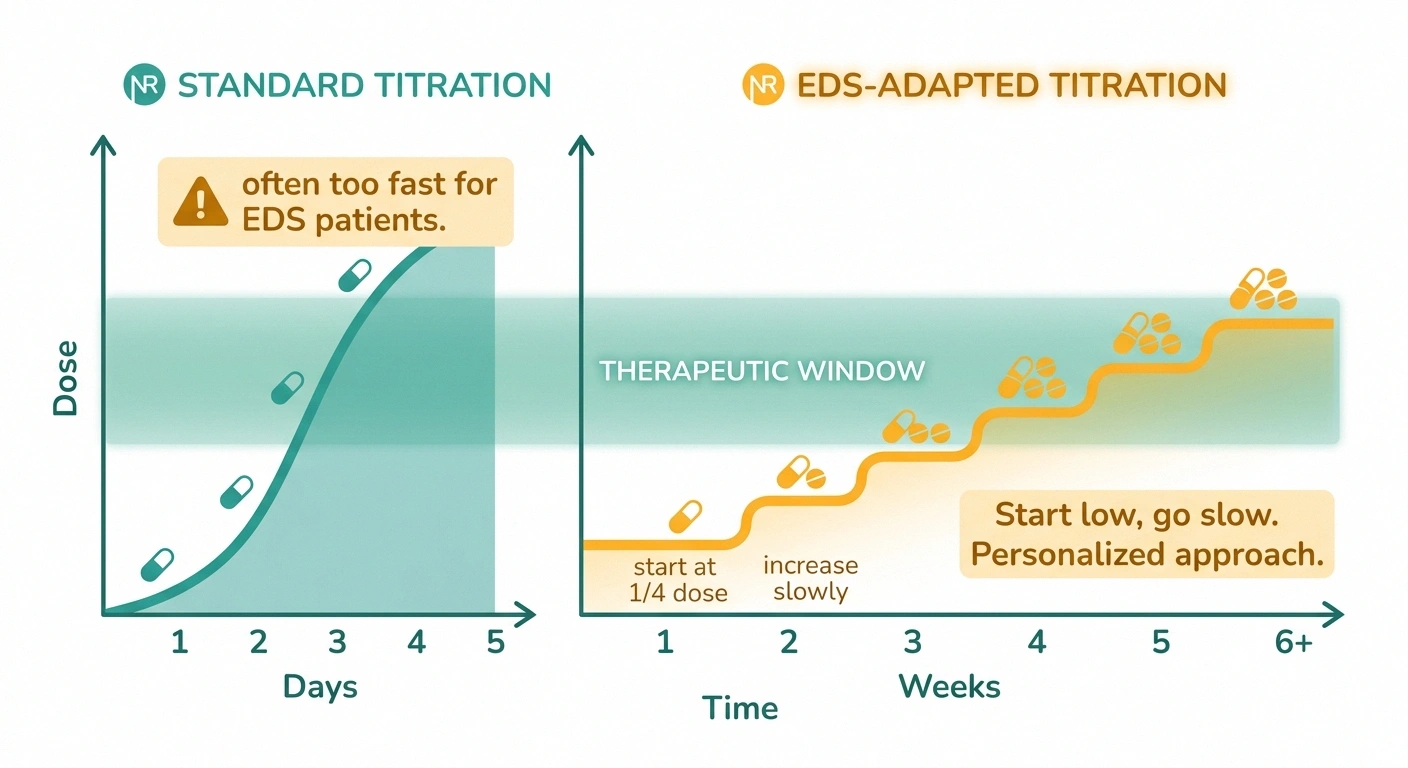
Many EDS patients cannot tolerate standard starting doses of gabapentin, pregabalin, or duloxetine. Start at a quarter of the printed dose and titrate up over weeks, not days. A drug labeled “didn't work” in an EDS patient may have been dosed too aggressively and abandoned before a tolerable therapeutic dose was reached.
The standard first-line neuropathic pain medications are gabapentin, pregabalin, and duloxetine. Gabapentin works well for many patients but can cause sedation, weight gain, and cognitive fog. Pregabalin is similar with sometimes better tolerability but similar side effects. Duloxetine is an SNRI that works on the pain signal from the other direction, and it has the advantage of also treating depression — which is relevant because chronic pain and EDS both have high rates of comorbid depression.
The modification for EDS is dose. Many EDS patients cannot tolerate the starting doses printed on the prescribing information. Starting at a quarter of the standard dose and titrating up slowly over weeks — rather than days — is a pattern experienced EDS-aware clinicians use. A patient who is told “this medication didn't work” may actually have been given a dose they couldn't tolerate long enough to feel the benefit.
Topical agents deserve a mention because they sidestep the drug-sensitivity issue. Topical capsaicin is the most studied topical for small fiber neuropathy — it causes a controlled depletion of substance P in local nerve endings. It burns when you first apply it, which is the mechanism at work. Lidocaine patches are gentler and work well for localized patches of burning pain.
Low-dose naltrexone (LDN) has emerged as an option for some small fiber neuropathy patients, particularly those with an inflammatory or immune-driven component. It's off-label for this use but well-tolerated, inexpensive, and worth discussing with a neurologist who has experience with it.
For a subset of patients — typically those with biopsy-confirmed SFN plus evidence of immune involvement — intravenous immunoglobulin (IVIG) is considered. It's expensive, not universally covered, and not first-line, but it can be a game-changer for the right patient.
The POTS and MCAS Overlap No One Warns You About
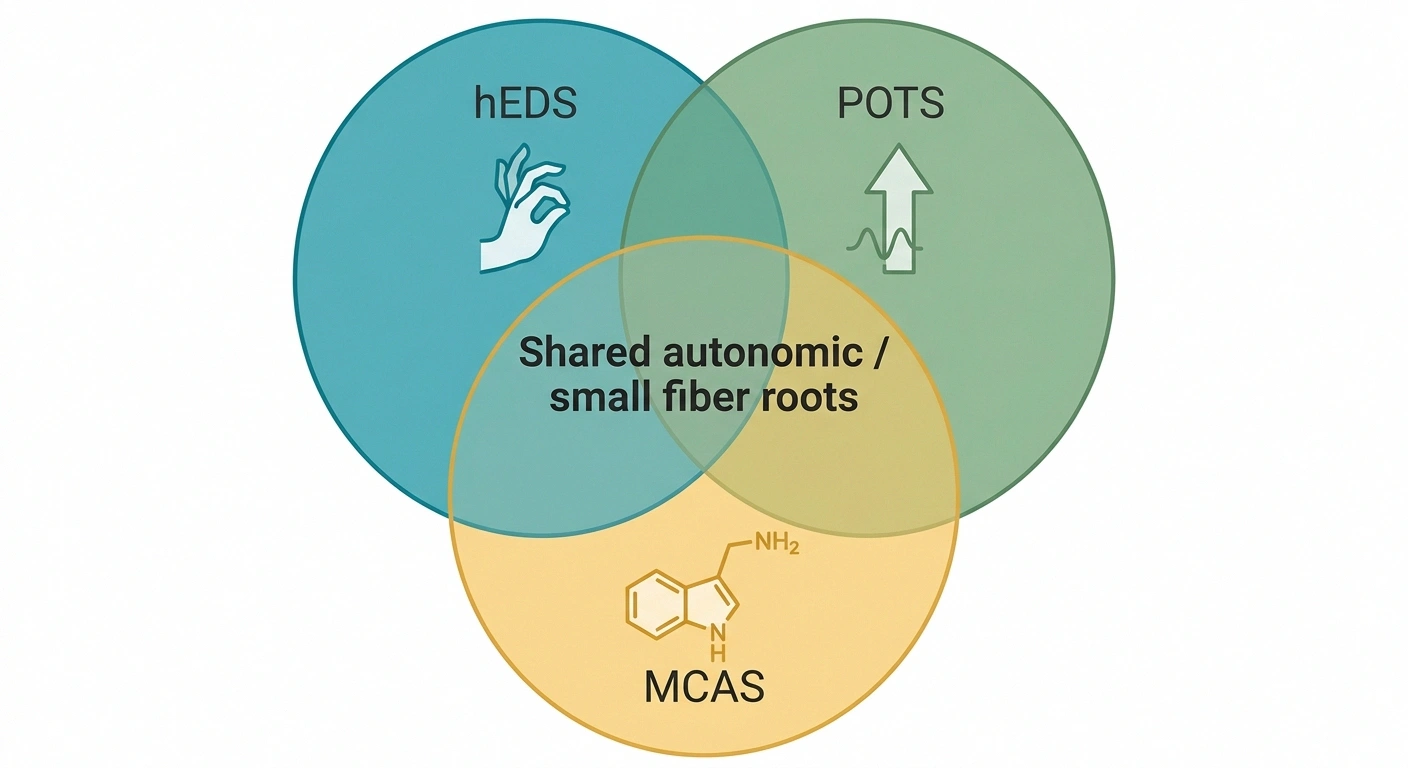
If you have hEDS and small fiber neuropathy, you may already know that postural orthostatic tachycardia syndrome (POTS) and mast cell activation syndrome (MCAS) show up in this population at rates far above the general average. This is not three unrelated problems piled up. They share a root in the autonomic nervous system and the small fiber network, and treating one sometimes improves the others.
The “Trifecta” — Shared Autonomic Roots
These three cluster together in hypermobile EDS at rates far above the general population. Treating one often improves the others — so if you have one, get evaluated for the other two.
POTS is diagnosed when your heart rate jumps more than 30 beats per minute within ten minutes of standing, without a corresponding drop in blood pressure. The small fibers that regulate blood vessel tone are involved — when they don't work properly, standing up causes blood to pool in the legs, the heart races to compensate, and the person feels lightheaded, fatigued, or foggy.
Treatment for POTS overlaps with autonomic small fiber treatment. Aggressive salt and fluid intake, compression garments, gradual exercise reconditioning, and sometimes medications like midodrine (a vasoconstrictor), fludrocortisone (a mineralocorticoid that expands blood volume), or beta-blockers for heart-rate control.
MCAS involves mast cells — immune cells — releasing histamine and other mediators inappropriately. Symptoms range from flushing and hives to GI distress to neurological symptoms. Treatment typically combines H1 antihistamines (cetirizine, fexofenadine), H2 blockers (famotidine), and mast cell stabilizers (cromolyn). In a patient with SFN plus MCAS, quieting the mast cells sometimes reduces the neuropathy flare pattern dramatically.
Joint Stability Work Still Matters
Treating the small fiber neuropathy does not mean ignoring the joints. Repeated mechanical trauma from subluxing joints is one of the contributing mechanisms we covered above, so joint stability work is still part of the plan — just no longer the whole plan.
Physical therapy specifically designed for hypermobility — not generic strengthening that assumes stable joints — builds the deep stabilizing muscles that your connective tissue can't. Programs like the Muldowney protocol for EDS are a starting point. Pool therapy, with the reduced joint load, is often well tolerated.
Bracing and compression support the joints and often incidentally improve circulation, which helps autonomic symptoms. Proper footwear matters disproportionately — shoes with good arch support and a stable heel counter reduce mechanical stress on the lower-extremity nerves.
Balance training earns a specific note. When you have both hypermobile joints and small fiber neuropathy, proprioception is doubly compromised. Falls risk is elevated, and deliberate balance work is worth building into your routine.
What Changes When You Have a Diagnosis
Getting the small fiber neuropathy diagnosis doesn't usually fix the symptoms overnight. What it does change is the frame of the problem. It converts “vague nerve pain my doctors don't take seriously” into an objectively documented condition with a known mechanism, a treatment literature, and — most importantly — a legitimate reason to build a specific treatment plan rather than grab-bag symptom management.
It also changes accommodations. An IENFD biopsy result in your chart is useful evidence for disability applications, workplace adjustments, school accommodations, and insurance coverage decisions. Many EDS patients describe the biopsy as the first piece of medical evidence that matched what they'd been saying for years.
And it changes the conversation with future doctors. “I have biopsy-confirmed small fiber neuropathy as part of my hypermobile EDS” is a sentence that opens doors a vaguer complaint does not.
When to Push Harder for Testing
Not every EDS patient with tingling needs a skin biopsy. But there are specific signals that should push you — or your doctor — to pursue the workup rather than chalk it up to the usual. Burning pain that interferes with sleep. Temperature sensitivity that feels disproportionate. Autonomic symptoms (heart rate swings, orthostatic dizziness, abnormal sweating) alongside the sensory complaints. Symptoms that have progressed over months, not just flared during a bad joint week.
If you see any of those signals and a general neurologist has told you your nerve conduction study is normal, the next step is either a skin biopsy request or a referral to a neurologist with small fiber neuropathy expertise. Academic medical centers and dedicated EDS clinics are your best bets. The physician specialty directory at The Ehlers-Danlos Society can help identify knowledgeable clinicians in your area.
And if you're having rapidly progressive weakness, new bladder or bowel dysfunction, or loss of function in a specific limb, that's not a routine investigation — that's an urgent evaluation. EDS or not, those are red-flag neurology findings.
A Note on Hope

I want to close with something that might sound small but isn't. For many EDS patients, reading the sentence “78% of hEDS patients have reduced IENFD consistent with small fiber neuropathy” is the first moment in years they feel believed. The damage is real. The pain is real. The mechanism makes sense. And there are options — imperfect, but real — that can meaningfully reduce the daily load.
The neuropathy may not be fully reversible; most small fiber neuropathies are chronic. But progression can usually be slowed, pain can usually be reduced, autonomic symptoms can usually be managed, and daily function can usually be improved. That's a different outcome from “learn to live with it with no explanation offered,” which is what too many EDS patients hear.
If you recognized yourself in this article, the next step is a conversation with a neurologist who will take the possibility of small fiber neuropathy seriously. Bring a list of your symptoms. Mention the IENFD skin biopsy by name. And bring a copy of one of the published studies — the medRxiv hEDS SFN paper, the Igharo 2023 European Journal of Neurology paper, or the Fernandez 2022 Journal of Internal Medicine paper — if your doctor isn't familiar with the literature. You'll be doing future EDS patients a favor even if the primary beneficiary is yourself.
Frequently Asked Questions
Does Ehlers-Danlos Syndrome cause neuropathy?
Research over the past decade has made the connection increasingly clear. Up to 78% of hypermobile EDS patients show reduced intraepidermal nerve fiber density on skin biopsy, which is the objective marker for small fiber neuropathy. The proposed mechanisms include collagen abnormalities in the nerve sheaths, microvascular fragility, repeated mechanical stress from hypermobile joints, and autoimmune overlap. Not every EDS patient has neuropathy, but the rate is dramatically higher than in the general population.
What is the difference between hEDS joint pain and nerve pain?
Joint pain from hypermobility is typically deep, aching, related to a specific joint, and worse with use or subluxation. Nerve pain — specifically small fiber neuropathy — is typically burning, electric, tingling, or a sunburn-like sensation. It's often diffuse or patchy rather than joint-localized, tends to worsen at rest and at night, and is often accompanied by temperature sensitivity and autonomic symptoms. Many patients have both, and untangling them is part of what a skilled clinician does.
How is small fiber neuropathy diagnosed in EDS?
Standard nerve conduction studies and EMG measure large myelinated fibers and are frequently normal in small fiber neuropathy. The gold standard is a 3mm skin punch biopsy from the distal leg, processed at a specialized lab that counts intraepidermal nerve fiber density. Complementary tests include QSART (sweat gland testing), tilt-table testing for autonomic involvement, and validated questionnaires like COMPASS-31 and SFN-SIQ. Ask specifically for a skin biopsy if a general neurologist tells you your nerve testing is normal but your symptoms persist.
What percentage of EDS patients have small fiber neuropathy?
The best-cited figure is about 78% of hypermobile EDS patients have reduced intraepidermal nerve fiber density consistent with small fiber neuropathy, from a 2022 study in the Journal of Internal Medicine. Other studies show somewhat different percentages depending on methodology and patient selection, but the pattern is consistent — a majority of hEDS patients have objective evidence of small fiber damage. This was not recognized for decades, which is why so many EDS patients spent years being told their nerve symptoms were psychological.
Why are EDS patients often sensitive to neuropathy medications?
EDS patients report atypical responses to many medications, including neuropathic pain drugs. Proposed reasons include altered drug metabolism in some subtypes, greater autonomic reactivity, higher baseline rates of mast cell activation (which amplifies side effects), and a tendency toward paradoxical reactions. The practical implication is that standard starting doses often need to be reduced significantly, with slow upward titration. A medication labeled “didn't work” in an EDS patient may actually have been dosed too aggressively and abandoned before a tolerable therapeutic dose was reached.
Can small fiber neuropathy in EDS be reversed?
Complete reversal is uncommon for most small fiber neuropathies, but the trajectory can often be slowed or stabilized, and symptoms can be substantially reduced with appropriate treatment. If there is a treatable driver — a B12 deficiency, an immune component responsive to IVIG, uncontrolled glucose, thyroid dysfunction — addressing it can produce meaningful improvement in IENFD over 12-24 months. For the specifically EDS-related component, the goal is usually stabilization and symptom management rather than cure, though this is an active research area.
Does POTS go with EDS neuropathy?
Yes, and frequently. POTS, small fiber neuropathy, and mast cell activation syndrome cluster together in hypermobile EDS at rates far above the general population. They share a root in autonomic dysfunction and small unmyelinated fiber pathology. Treatment for one often helps the others — aggressive fluids and salt, compression garments, and gradual reconditioning benefit both POTS and autonomic small fiber neuropathy. If you have one of these diagnoses, it is worth being evaluated for the other two.
What kind of doctor should I see for EDS-related neuropathy?
The ideal is a neurologist with specific small fiber neuropathy expertise, ideally at an academic medical center or a clinic that sees EDS patients regularly. Autonomic neurologists are a subset especially worth seeking out if you have orthostatic symptoms alongside the burning pain. The Ehlers-Danlos Society maintains a physician directory that can help. General neurologists without SFN experience sometimes dismiss normal nerve conduction as the end of the workup — a pattern worth being aware of so you can advocate for the right next test.