
When my friend Margaret finally got her diagnosis after four years of progressive numbness, weakness, and what her doctors called “idiopathic neuropathy,” we both sat in stunned silence. Amyloid neuropathy — specifically a type called ATTR amyloidosis — wasn't even on our radar. Neither of us had heard of it. And yet, once the diagnosis landed, it changed everything: there were treatments. Real, approved, science-backed treatments that could slow the disease in its tracks.
That experience — and the years of unnecessary suffering Margaret went through before anyone connected the dots — is exactly why I want to talk about ATTR amyloid neuropathy today. It's rare, yes. But it's far less rare than most people think, and it's one of the most important diagnoses not to miss. If you or someone you love has been struggling with progressive neuropathy that doesn't fit the usual patterns, or if your doctor has used the word “idiopathic” more times than you can count, this article is for you.
What Is ATTR Amyloid Neuropathy?
ATTR stands for transthyretin amyloidosis — a condition where a protein called transthyretin (TTR) misfolds and clumps together into sticky, abnormal deposits called amyloid fibrils. These deposits build up in tissues throughout the body, including the peripheral nerves, heart, kidneys, and eyes.
ATTR amyloid neuropathy is caused by a misfolded protein — transthyretin (TTR) — that deposits in peripheral nerves. It affects an estimated 50,000 people worldwide with hereditary forms alone, and is significantly underdiagnosed.
Transthyretin is a normal protein produced mainly by the liver. Its job is to carry thyroid hormone and vitamin A (retinol) through the bloodstream. Under normal circumstances, it folds into the right shape and does its job without incident. But in ATTR amyloidosis, the protein becomes unstable — either because of a genetic mutation or simply because of the aging process — and it begins to unfold and aggregate into those damaging fibrils.
When these deposits accumulate in the peripheral nerves, the result is ATTR polyneuropathy (ATTR-PN). When they primarily accumulate in the heart, it's ATTR cardiomyopathy (ATTR-CM). Many patients have both.
ATTR neuropathy is classified as a type of peripheral neuropathy, and it shares symptoms with many other forms. Understanding the stages of neuropathy can help you recognize how this disease progresses — and why catching it early matters so much.
The Two Forms: Hereditary and Wild-Type ATTR
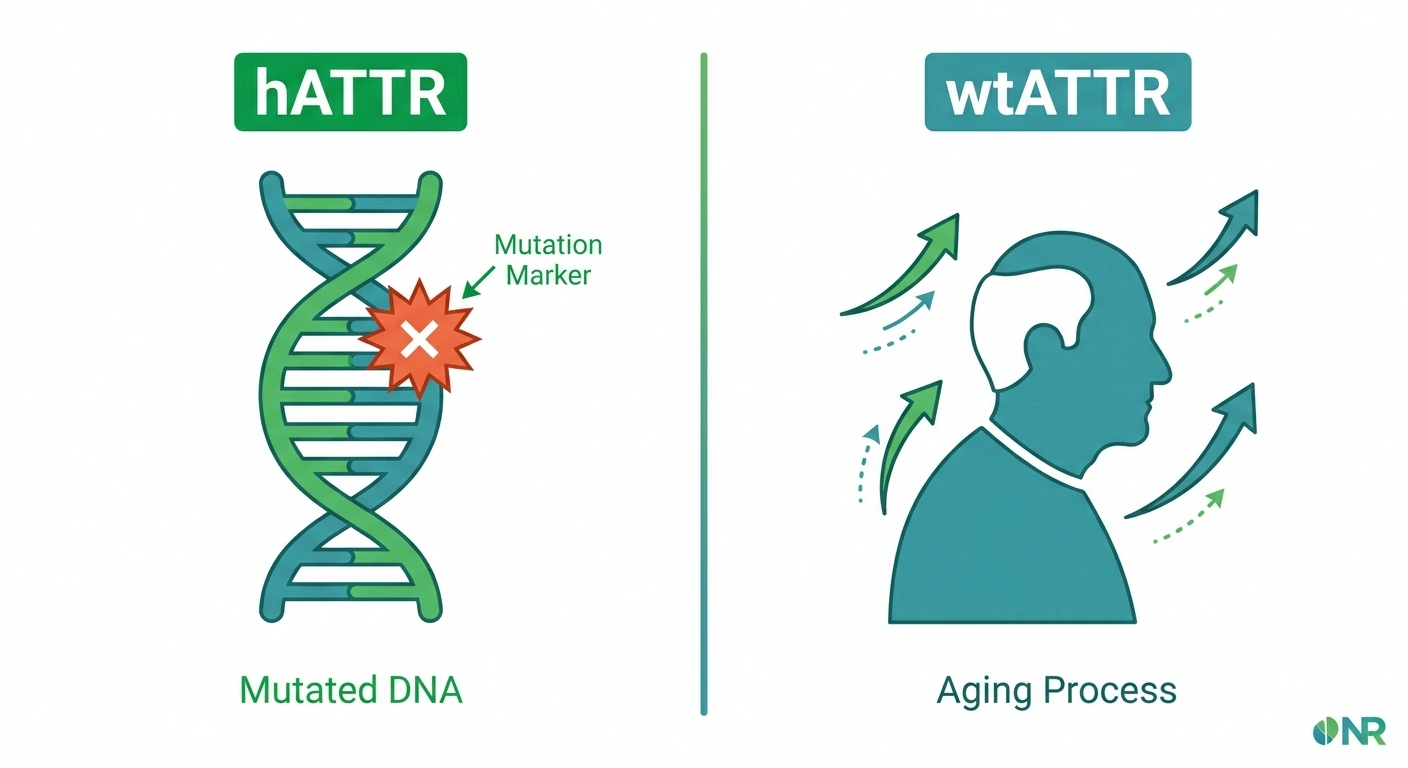
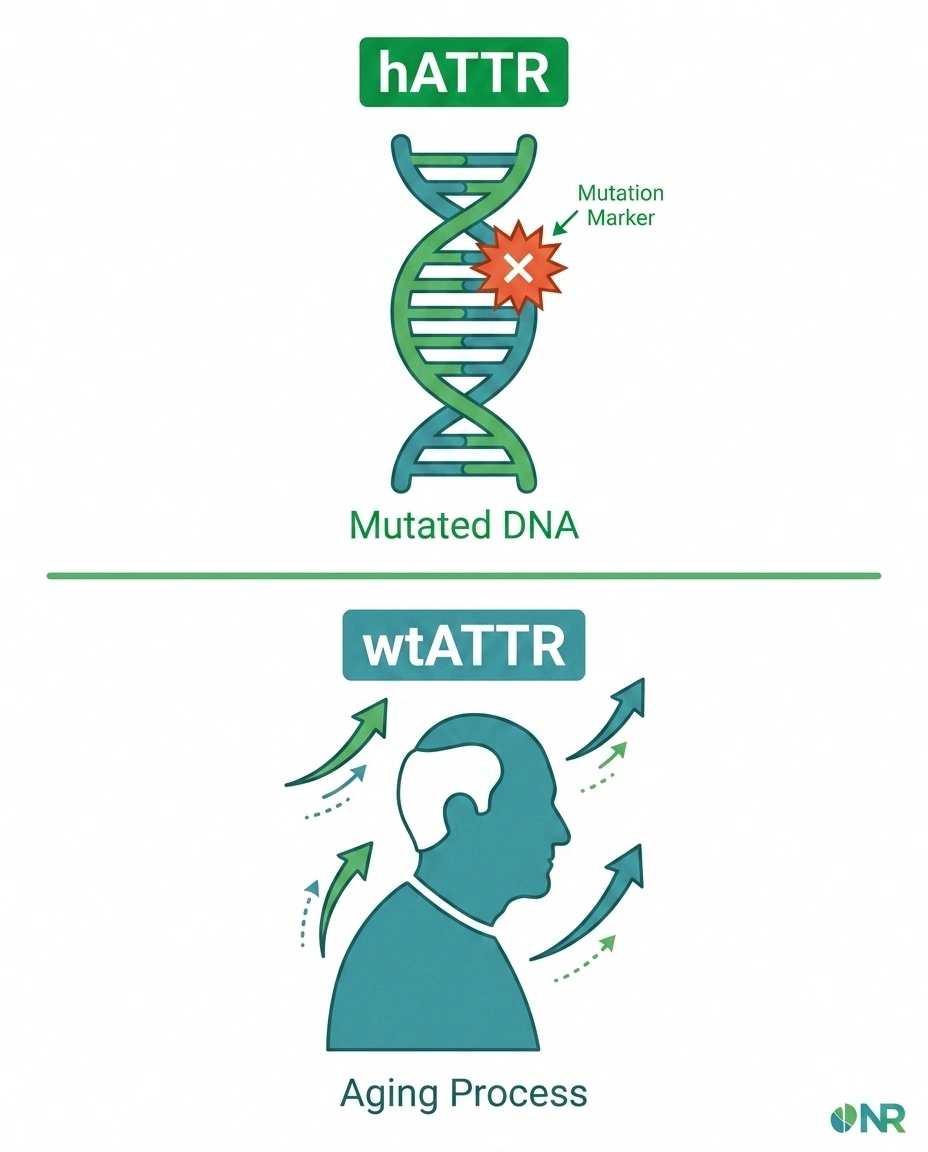
ATTR amyloidosis comes in two distinct varieties, and understanding the difference is key to knowing your risk.
- Caused by TTR gene mutation
- Autosomal dominant (50% inheritance risk)
- Often onset in 30s–60s
- Common: Val30Met, Val122Ile, Thr60Ala
- Mainly polyneuropathy ± cardiac
- No genetic mutation — age-related
- Not inherited
- Primarily men over 60
- Normal TTR gene
- Mainly cardiac ± neuropathy/CTS
Hereditary ATTR (hATTR) is caused by a mutation in the TTR gene. More than 100 different mutations have been identified, but a few are more common than others:
- Val30Met (V30M): The most common mutation worldwide, particularly prevalent in Portugal, Sweden, and Japan. This variant primarily causes polyneuropathy and often presents in younger adults (30s–40s).
- Val122Ile (V122I): More common in people of West African descent; roughly 3–4% of African Americans carry this variant. It predominantly affects the heart but can also cause neuropathy.
- Thr60Ala (T60A): Found more frequently in Irish and British populations; typically causes combined cardiac and neurological involvement.
Because hATTR is hereditary, first-degree relatives of someone diagnosed with it have a 50% chance of carrying the mutation. Genetic testing is strongly recommended for family members after a diagnosis.
Wild-type ATTR (wtATTR) is not hereditary — it develops from the normal aging process. For reasons that aren't fully understood, the normal (non-mutated) TTR protein becomes unstable over time in some older adults and begins to aggregate. Wild-type ATTR primarily affects the heart, but peripheral neuropathy and carpal tunnel syndrome are also common. It predominantly affects men over the age of 60.
Wild-type ATTR was once thought to be exceedingly rare, but better diagnostic tools have revealed it's actually quite common in older patients — present in up to 25% of people over 80 at autopsy, though not all had clinical symptoms.
Why ATTR Is So Often Missed
Here's the painful truth: ATTR neuropathy is significantly underdiagnosed. Studies suggest that the average time from symptom onset to correct diagnosis is five to seven years. That's five to seven years of progressive nerve damage that, with modern treatments, could have been slowed or stopped.
The average time from ATTR symptom onset to correct diagnosis is 5–7 years. During this time, nerve damage continues. ATTR often masquerades as idiopathic neuropathy or CIDP. If your neuropathy is progressive and unexplained, ask your neurologist specifically about ATTR testing.
The reasons for this diagnostic delay are understandable. ATTR neuropathy mimics several other conditions:
- It looks like CIDP (chronic inflammatory demyelinating polyneuropathy) — and patients are sometimes treated with IVIG for years before the real diagnosis is made
- It presents similarly to idiopathic neuropathy, particularly in older adults
- Carpal tunnel syndrome — often an early manifestation — is treated as carpal tunnel without further investigation
- Autonomic symptoms (dizziness, GI problems, bladder issues) may be attributed to other causes
Awareness is the first step to closing this gap. If you've received a diagnosis of “idiopathic” neuropathy and your symptoms are progressing, asking your neurologist about ATTR testing is entirely reasonable.
Red Flags That Should Trigger ATTR Testing
Certain clinical features raise the suspicion for ATTR significantly. Neurologists and amyloid specialists look for a constellation of findings — what some call the “ATTR phenotype”:
A 2024 expert panel in Muscle & Nerve recommends genetic testing and biopsy for any progressive neuropathy with autonomic involvement, bilateral carpal tunnel, or cardiac thickening — especially when standard treatments aren't working. Earlier testing = earlier treatment = better outcomes.
- Bilateral carpal tunnel syndrome before age 60, especially if it required surgery
- Progressive neuropathy in an older adult without a clear cause (diabetes, alcohol, vitamin deficiency, toxic exposure)
- Family history of neuropathy, heart failure, or unexplained progressive illness
- Combined neuropathy and heart involvement — an enlarged or thickened heart alongside neuropathy is a significant red flag
- Autonomic dysfunction — especially early and prominent autonomic symptoms (orthostatic hypotension, GI dysmotility, erectile dysfunction)
- Lumbar spinal stenosis requiring surgery, especially if followed by neuropathy
- Vitreous opacities (deposits in the eye that affect vision)
- Neuropathy that doesn't respond to typical treatments and continues to progress
None of these features alone confirms ATTR, but any combination should prompt a conversation with your neurologist about further testing. The conditions that affect the nerves are complex, which is why a thorough neuropathy diagnosis process is so important.
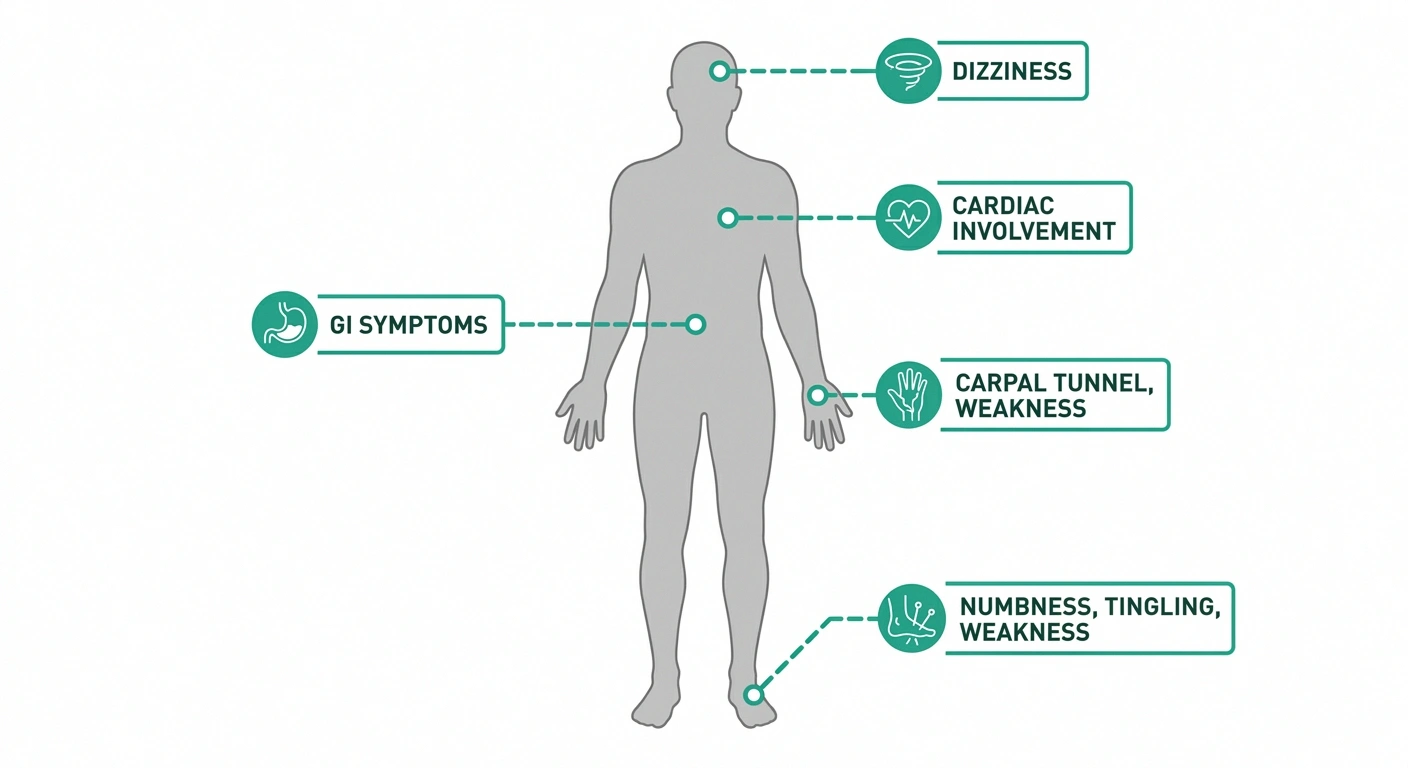
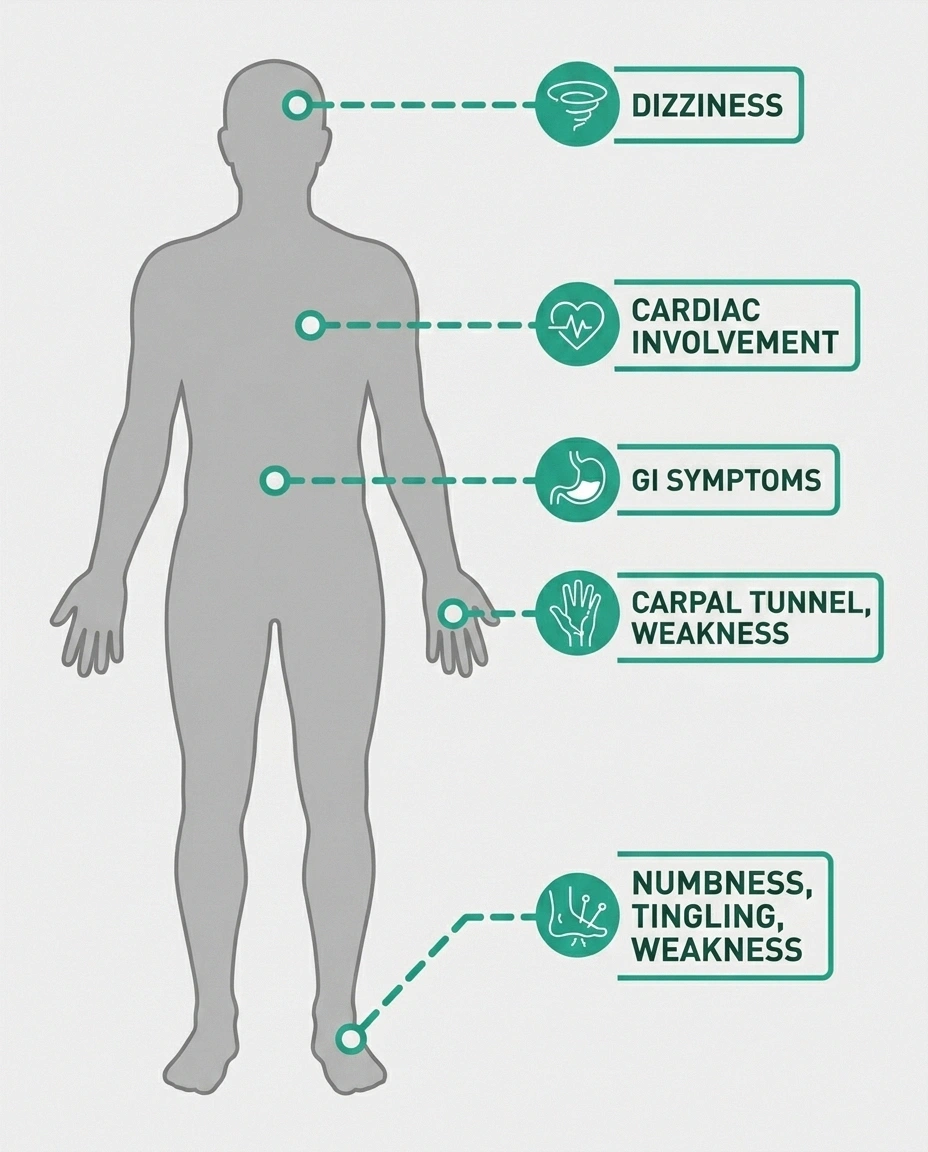
Recognizing the Symptoms of ATTR Polyneuropathy
ATTR polyneuropathy typically starts in the lower extremities and progresses upward — what neurologists call a length-dependent pattern. This means the longest nerve fibers are affected first, which is why the feet and lower legs are usually involved before the hands.
Sensory symptoms often come first:
- Numbness, tingling, or burning in the feet and lower legs
- Pain — which can range from mild discomfort to severe, burning neuropathic pain
- Loss of temperature sensation (often early and prominent in ATTR)
- Loss of pain sensation, which creates risk for unnoticed injuries
- Eventually, loss of vibration and position sense (proprioception)
Motor symptoms develop as the disease progresses:
- Muscle weakness in the legs and feet
- Difficulty walking or climbing stairs
- Foot drop (difficulty lifting the front of the foot)
- Weakness in the hands and arms in later stages
One important distinction: in ATTR neuropathy, small fiber involvement (temperature and pain sensation) tends to be prominent and early, which can help distinguish it from some other forms of neuropathy.
Autonomic Symptoms: The Hidden Dimension
One of the features that makes ATTR neuropathy particularly distinctive — and particularly challenging — is the degree to which it can affect the autonomic nervous system. Autonomic neuropathy affects the nerves that control involuntary body functions, and in ATTR, these symptoms can be severe and significantly impact quality of life.
Common autonomic manifestations of ATTR include:
- Orthostatic hypotension: A significant drop in blood pressure when standing, causing dizziness, lightheadedness, or fainting
- Gastrointestinal dysmotility: Alternating constipation and diarrhea, early satiety, nausea, weight loss — often one of the most disabling features
- Bladder dysfunction: Urgency, frequency, urinary retention, or incontinence
- Sexual dysfunction: Erectile dysfunction is common in men with ATTR
- Sweating abnormalities: Reduced or absent sweating (anhidrosis), sometimes followed by compensatory excessive sweating in unaffected areas
- Cardiac arrhythmias: Relating to the cardiac involvement that often accompanies the neuropathy
The GI symptoms in particular can be profoundly debilitating — patients may lose 20–30 pounds or more as the disease progresses without treatment, and the roller-coaster of constipation and diarrhea can dominate daily life.
How ATTR Neuropathy Is Diagnosed
Diagnosing ATTR requires combining clinical suspicion with specific diagnostic tests. Here's what the workup typically looks like:
Nerve conduction studies and EMG confirm the presence of polyneuropathy and help characterize it. ATTR typically causes an axonal polyneuropathy (damage to the nerve fibers themselves rather than the myelin sheath), though this isn't universal.
Genetic testing — a blood test — is done to look for TTR gene mutations. This confirms hereditary ATTR and identifies the specific variant. Importantly, a negative genetic test does not rule out ATTR, since wild-type ATTR has no mutation.
Tissue biopsy is the definitive diagnostic step. When amyloid deposits are found in tissue (often abdominal fat pad, salivary gland, or nerve tissue), they can be identified by Congo red staining. Critically, mass spectrometry is now used to type the amyloid — confirming that the deposits are TTR-type and not one of the other forms of amyloidosis (like AL amyloidosis, which is treated completely differently).
Cardiac evaluation — typically an echocardiogram and cardiac MRI — is important because many ATTR patients have cardiac involvement, and the heart findings (concentric thickening, “sparkling” appearance on echo, specific MRI patterns) can support the diagnosis and guide management.
Technetium pyrophosphate (99mTc-PYP) scan is a nuclear medicine scan that can detect TTR cardiac deposits with high sensitivity and specificity. It's now widely used as part of the diagnostic workup, especially when cardiac involvement is suspected.
A Revolution in Treatment: Modern Options for ATTR
This is where the story of ATTR becomes genuinely hopeful. In the past decade, treatment of ATTR has been transformed. Where once there were no approved therapies and a liver transplant was the only real option, patients now have access to multiple effective medications that can dramatically slow — and in some cases stabilize — the disease.
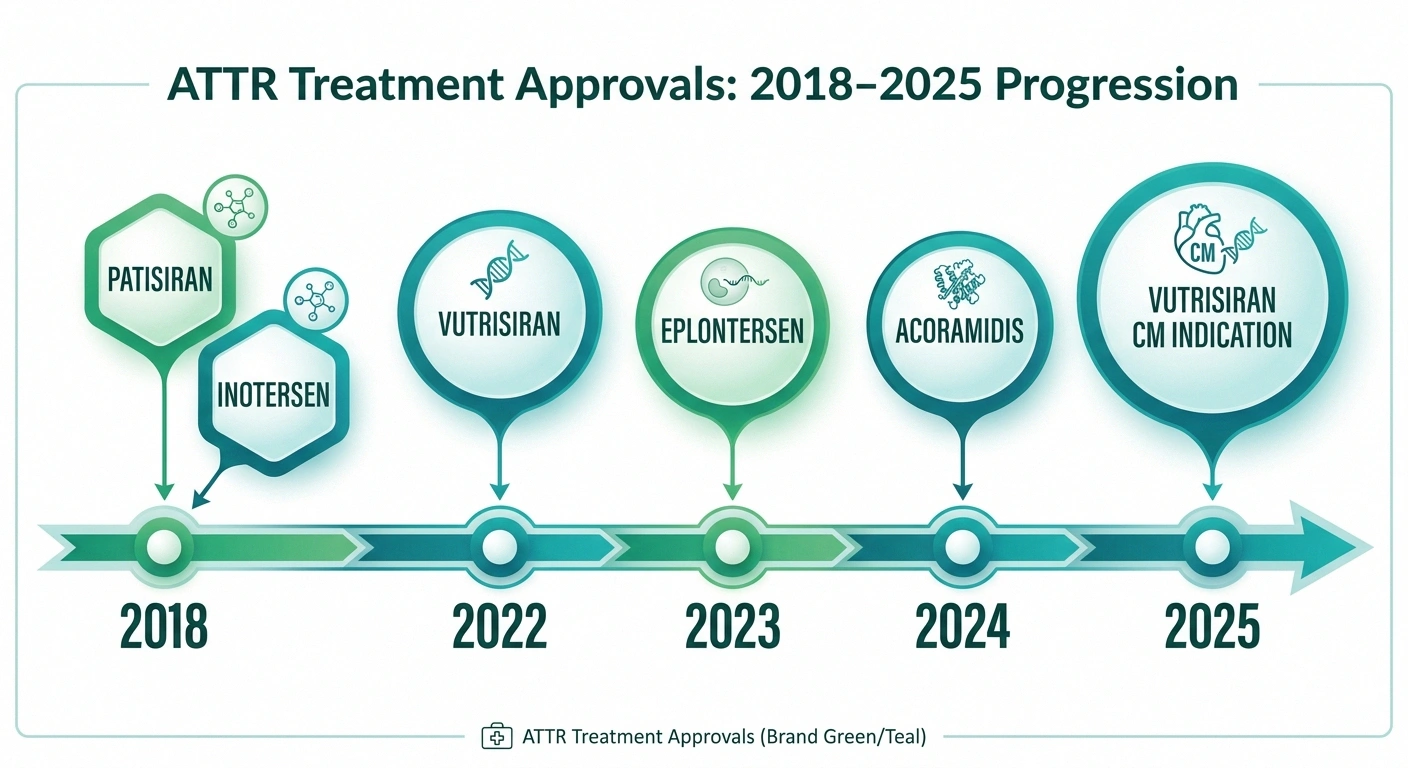
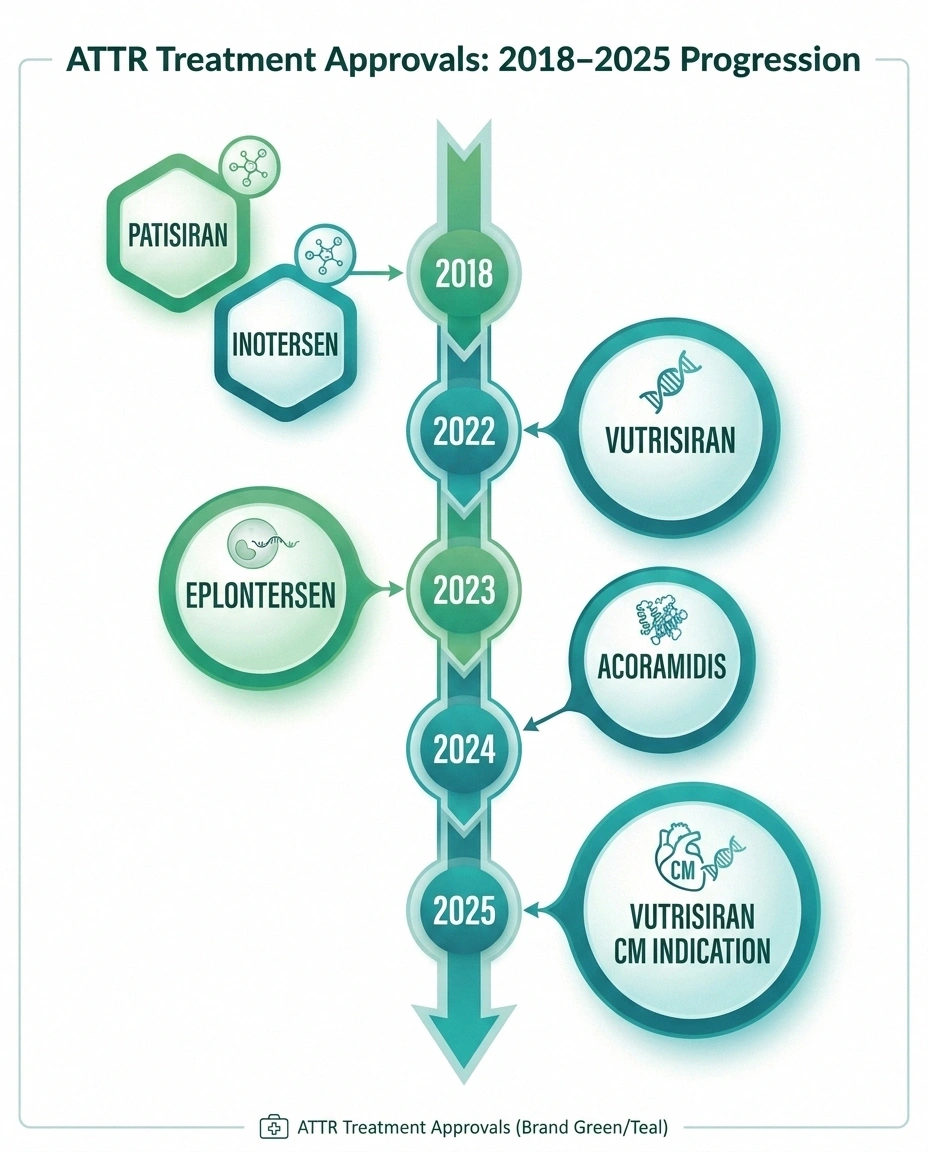
| Drug | Type | How Given | Approved For |
|---|---|---|---|
| Patisiran (Onpattro) | RNAi | IV every 3 weeks | hATTR-PN |
| Vutrisiran (Amvuttra) | RNAi | Subcut. every 3 months | hATTR-PN + ATTR-CM |
| Eplontersen (Wainua) | ASO | Subcut. monthly | hATTR-PN |
| Inotersen (Tegsedi) | ASO | Weekly injection | hATTR-PN |
| Tafamidis / Acoramidis | Stabilizer | Oral daily | ATTR-CM (+ some PN) |
Treatments work through different mechanisms:
TTR Stabilizers work by stabilizing the TTR protein so it doesn't unfold and aggregate. Tafamidis (Vyndaqel, Vyndamax) is the most widely used stabilizer, primarily approved for ATTR cardiomyopathy but also used for the polyneuropathy in some contexts. Acoramidis (received FDA approval in November 2024) is a newer, more selective stabilizer also approved for ATTR-CM in adults.
Gene-Silencing Therapies represent the biggest advance. These drugs work by reducing TTR production in the liver, thereby dramatically cutting the amount of protein available to misfold:
- Patisiran (Onpattro) — the first RNA interference (RNAi) drug approved for a neurological disease. Given as an IV infusion every 3 weeks, it reduces TTR levels by approximately 80%. Approved for hATTR with polyneuropathy.
- Vutrisiran (Amvuttra) — a subcutaneous injection given every 3 months, approved for hATTR-PN. As of March 2025, also approved for wild-type or variant ATTR cardiomyopathy in adults.
- Inotersen (Tegsedi) — a weekly subcutaneous injection, approved for hATTR-PN. Works through antisense oligonucleotide (ASO) technology.
- Eplontersen (Wainua) — a newer ASO given once monthly. In clinical trials, it reduced serum TTR by 81.7% at week 65, with significant improvements in neuropathy impairment scores and quality of life.
Current expert consensus recommends gene-silencing therapies as first-line treatment for ATTR polyneuropathy, given their superior TTR reduction and strong clinical trial data. The key message is that earlier treatment is better — these drugs slow progression but cannot reverse established nerve damage.
Liver transplantation was the only disease-modifying option before drugs became available. Because the liver produces ~90% of circulating TTR, replacing the liver with a healthy one dramatically reduces TTR production. Transplantation is still used in some cases, particularly in younger patients with the Val30Met mutation, but it has largely been superseded by drug therapy.
The research into nerve regeneration and repair is also advancing rapidly. For context on where that science stands, see our article on nerve regeneration research and the path toward a cure.
ATTR vs. Other Rare Neuropathies: How to Tell the Difference
ATTR can look remarkably like other forms of inherited neuropathy. Hereditary neuropathy (CMT disease) is a common consideration, particularly in younger patients with a family history of neuropathy. Key differences:
- CMT typically causes a demyelinating pattern on nerve conduction studies; ATTR is usually axonal
- ATTR has significant autonomic involvement; CMT typically does not
- ATTR often causes cardiac involvement; CMT does not
- ATTR can have late onset even in hereditary forms; CMT typically presents in childhood or early adulthood
ATTR is also confused with CIDP, particularly because both are progressive, can be treatment-resistant, and can show similar nerve conduction changes. The key red flag is CIDP that doesn't respond to IVIG treatment or keeps relapsing — this should trigger ATTR testing.
Living Well with ATTR Neuropathy
Managing ATTR neuropathy goes beyond disease-modifying therapy. Symptom management is an important part of care, particularly for autonomic symptoms:

ATTR neuropathy is no longer untreatable. Gene-silencing therapies can dramatically slow progression when started early. The key is not missing the diagnosis — and that means knowing the red flags and advocating for the right tests.
- Orthostatic hypotension: Increasing fluid and salt intake, compression stockings, elevating the head of the bed, and medications (midodrine, fludrocortisone) can help manage dizziness and falls
- GI symptoms: Working with a gastroenterologist, dietary modifications (smaller and more frequent meals, low-fat diet for dysmotility), and medications for specific symptoms
- Pain management: Same medications used for other forms of neuropathic pain — gabapentin, pregabalin, duloxetine, TCAs — though responses vary
- Fall prevention: Especially important as balance and proprioception are affected; home modifications, physical therapy, and appropriate assistive devices
- Nutrition: Weight loss can be severe; working with a dietitian is important, particularly for patients with significant GI involvement
Physical therapy plays an important role in maintaining function and preventing complications. Working with a PT experienced in neuropathy can help maintain strength and mobility even as the disease progresses.
Emotional support is equally vital. ATTR neuropathy is life-changing, and the journey — from diagnostic odyssey to treatment — takes an enormous toll. Connecting with others through organizations like the Amyloidosis Research Consortium and patient support communities can provide both information and a sense of not being alone.
The Importance of a Specialized Center
ATTR amyloidosis is complex enough that care at a specialized amyloid center — or at minimum, a neurologist with experience in the condition — makes a significant difference. These centers have:
- Experience interpreting complex diagnostic results
- Multidisciplinary teams (neurology, cardiology, gastroenterology, genetics)
- Access to newer treatments and clinical trials
- Coordination of care that individual specialists in community settings may not provide
The National Amyloidosis Centre (UK) and multiple academic medical centers in the US maintain dedicated ATTR clinics. Your neurologist can help with a referral, or you can search through the Amyloidosis Research Consortium website for centers near you.
A Message of Hope
When Margaret got her diagnosis, we both felt something unexpected alongside the fear: relief. After years of uncertainty, there was finally a name, an explanation, and — most importantly — options. The treatments available today for ATTR neuropathy are extraordinary compared to what existed just ten years ago. Patients who start treatment early can maintain function and quality of life for many years.
ATTR amyloidosis is no longer a sentence. It's a condition with a treatment plan. And that changes everything.
If you think you or a family member might have ATTR — if something in this article has resonated with you — please talk to your neurologist. Ask specifically about ATTR testing. Be persistent. The stakes are high, but so is the potential for meaningful help.
Frequently Asked Questions
What is the difference between ATTR and AL amyloidosis?
ATTR amyloidosis is caused by misfolded transthyretin protein, while AL amyloidosis is caused by misfolded immunoglobulin light chain proteins produced by abnormal plasma cells. They look similar under a microscope but are caused by completely different diseases and require completely different treatments. Mass spectrometry of biopsy tissue is the most reliable way to distinguish them. AL amyloidosis is treated with chemotherapy targeting the plasma cell disorder; ATTR is treated with TTR-targeting drugs.
Is amyloid neuropathy hereditary?
Hereditary ATTR (hATTR) is inherited in an autosomal dominant pattern, meaning a person needs only one copy of the mutated gene from one parent to be at risk. First-degree relatives of a person with hATTR have a 50% chance of carrying the mutation. However, having the mutation does not guarantee developing the disease — penetrance varies by mutation and other factors. Wild-type ATTR is not hereditary.
How is ATTR amyloidosis diagnosed?
Diagnosis involves a combination of genetic testing (blood test for TTR mutations), tissue biopsy with Congo red staining and mass spectrometry to confirm TTR-type amyloid deposits, nerve conduction studies to characterize the neuropathy, and cardiac evaluation (echocardiogram, cardiac MRI, nuclear medicine scan) to assess for cardiac involvement. The complete workup usually requires a specialist experienced in amyloid diseases.
What are the first signs of ATTR amyloidosis?
Early signs often include bilateral carpal tunnel syndrome (which may occur years before neuropathy), early sensory symptoms in the feet such as numbness or burning, and autonomic symptoms like dizziness when standing or gastrointestinal problems. Some patients develop lumbar spinal stenosis requiring surgery as an early manifestation. The combination of carpal tunnel, neuropathy, and autonomic or cardiac symptoms together should prompt ATTR evaluation.
Can ATTR neuropathy be treated?
Yes. Multiple FDA-approved disease-modifying treatments are now available, including RNA interference drugs (patisiran, vutrisiran), antisense oligonucleotides (inotersen, eplontersen), and TTR stabilizers (tafamidis, acoramidis). These drugs significantly slow or halt disease progression. The key is early treatment — established nerve damage cannot be reversed, but ongoing damage can be stopped. This is why early diagnosis is so critical.
How long can you live with ATTR amyloidosis?
Life expectancy depends on when the disease is diagnosed and treated, which variant is involved, and the degree of cardiac involvement. Untreated ATTR polyneuropathy typically progresses to wheelchair dependence within 10 years of symptom onset, with survival often affected by cardiac and autonomic complications. With modern disease-modifying therapy started early, progression can be substantially slowed and quality of life maintained much longer. Patients treated early with effective therapies can live for many years with stable or minimally progressing disease.
What is the difference between ATTR-PN and ATTR-CM?
ATTR-PN (polyneuropathy) refers to the nervous system manifestation — damage to the peripheral nerves causing numbness, weakness, and autonomic dysfunction. ATTR-CM (cardiomyopathy) refers to cardiac involvement — thickening of the heart walls leading to heart failure and arrhythmias. Many patients with ATTR have both. Different drugs have been specifically approved for one or both indications, and management requires both a neurologist and cardiologist working together.


