
CIDP Explained: Chronic Inflammatory Demyelinating Polyneuropathy
I still remember the first message I received from a woman named Carol. She'd been losing strength in her legs for months — slowly, quietly, in a way that made her think she was just getting older. By the time a neurologist finally mentioned CIDP, she'd spent nearly a year wondering what was wrong. “Why didn't anyone catch this sooner?” she wrote. It's a question I hear far too often.
Chronic Inflammatory Demyelinating Polyneuropathy — CIDP for short — is one of those conditions that hides in plain sight. It's rare enough that many doctors don't think of it right away, but common enough that I've connected with hundreds of people living with it. If you've recently been diagnosed or you're still searching for answers, this guide is for you.
What Is CIDP?
CIDP is a rare autoimmune disorder that attacks the myelin sheath — the protective insulation wrapped around your peripheral nerves. Think of myelin like the rubber coating on an electrical wire. When your immune system strips it away, nerve signals slow down or get lost entirely. The result is progressive weakness, numbness, and sometimes pain that develops over at least eight weeks.
According to the GBS-CIDP Foundation International, new cases occur at a rate of about 1 to 2 per 100,000 people per year, though prevalence may be as high as 9 per 100,000 because the condition often goes undiagnosed for months or years. CIDP is more common in men than women and can begin at any age, though it's most frequently diagnosed between ages 40 and 60.
If you're familiar with Guillain-Barré Syndrome (GBS), think of CIDP as its chronic cousin. GBS strikes suddenly and typically resolves within weeks to months. CIDP develops more gradually and can persist for years — sometimes in a relapsing-remitting pattern, sometimes as a slow, steady decline.
How CIDP Differs from Other Types of Neuropathy
Most types of neuropathy damage the nerve fiber itself — the axon. That's what happens in diabetic neuropathy, chemotherapy-induced neuropathy, and most other forms. CIDP is fundamentally different because it's a demyelinating neuropathy, meaning the damage targets the myelin coating rather than the nerve core.
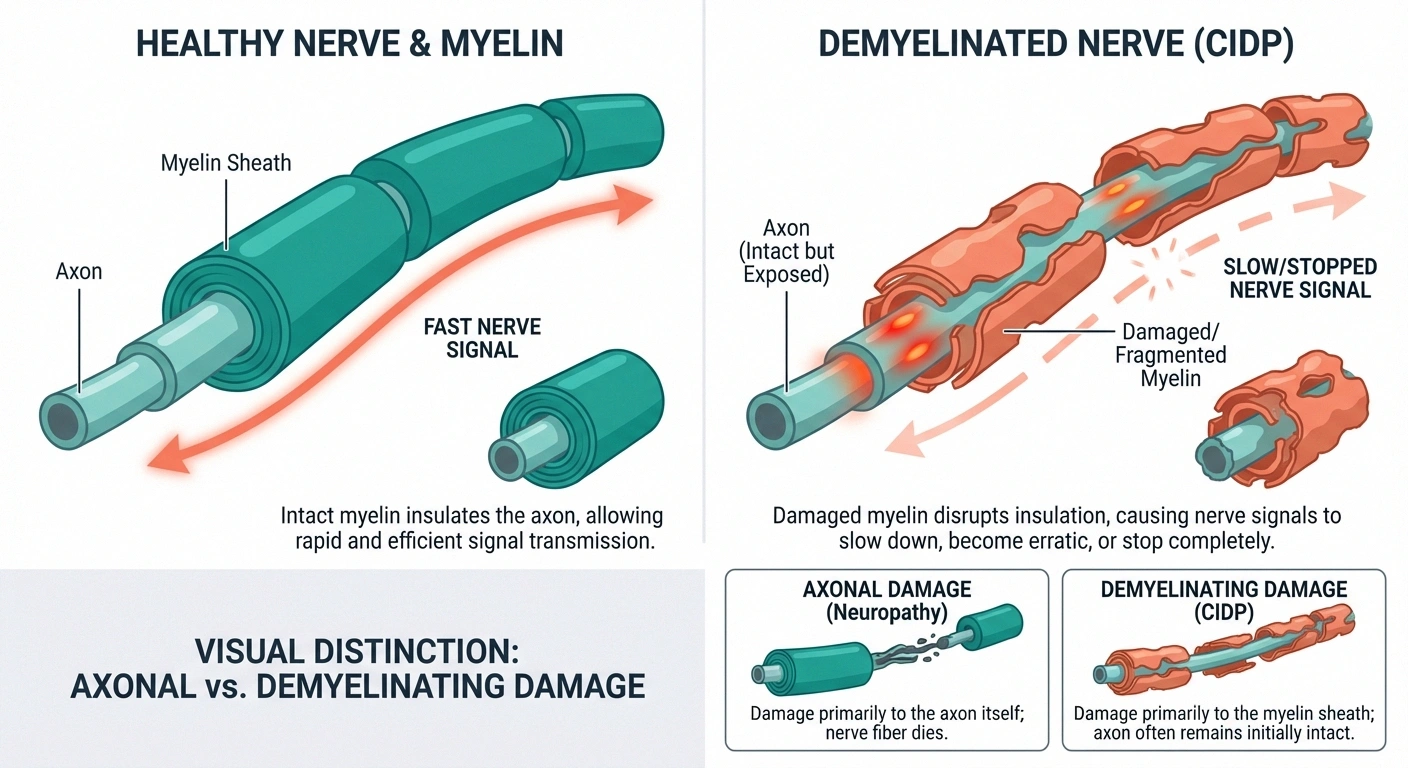
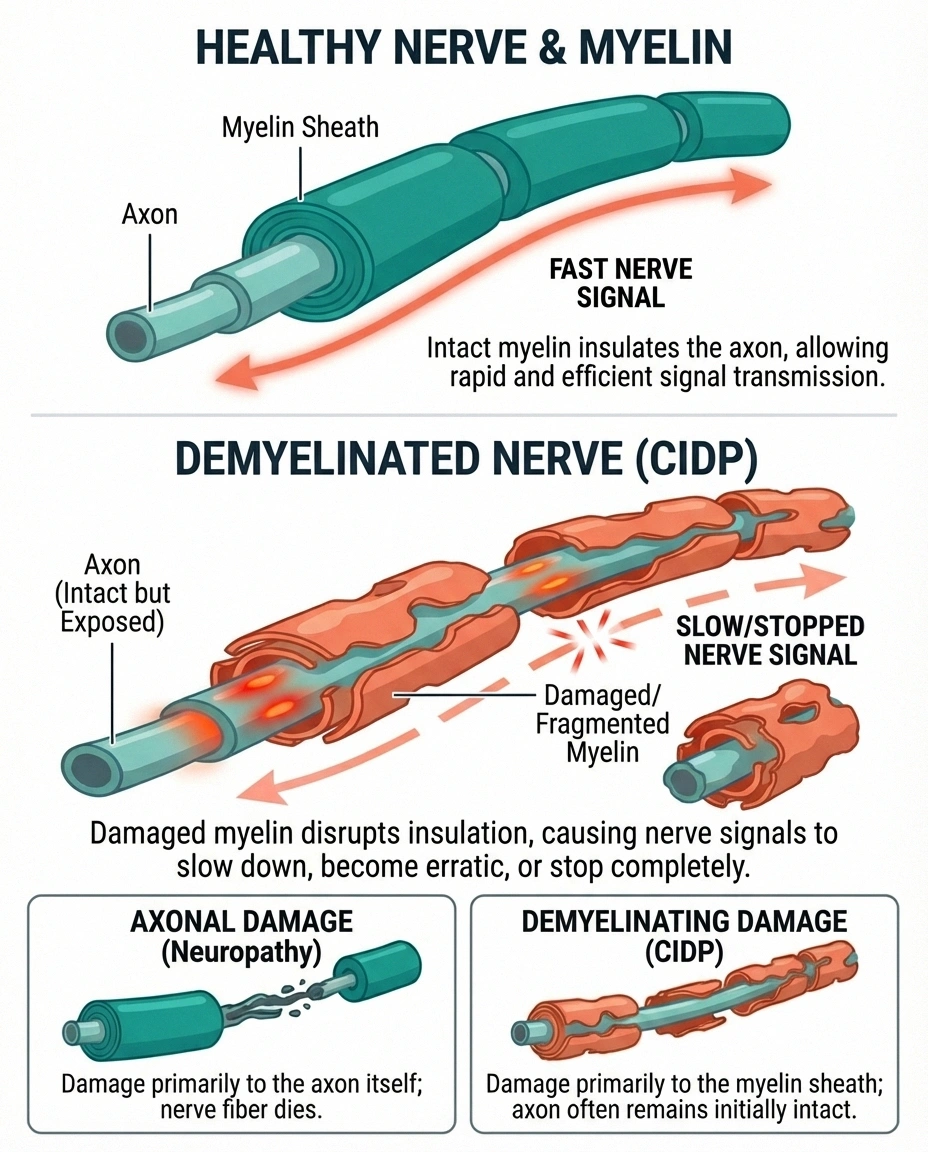
This distinction matters for two important reasons. First, demyelinated nerves can often be repaired if treatment begins early enough — the body can rebuild myelin in ways it can't easily regenerate destroyed axons. Second, because CIDP is immune-mediated, the treatments are completely different from standard neuropathy medications. Gabapentin and pregabalin may help manage symptoms, but they don't address the underlying immune attack.
Understanding whether your neuropathy is polyneuropathy versus mononeuropathy and whether it's demyelinating or axonal is one of the most important diagnostic distinctions your neurologist will make. It changes everything about your treatment plan and your prognosis.
Symptoms of CIDP: What to Watch For
The hallmark of CIDP is symmetrical weakness that develops gradually over at least eight weeks. Unlike many neuropathies that start in the feet, CIDP often affects both proximal muscles (hips, thighs, shoulders) and distal muscles (hands, feet) — sometimes simultaneously.
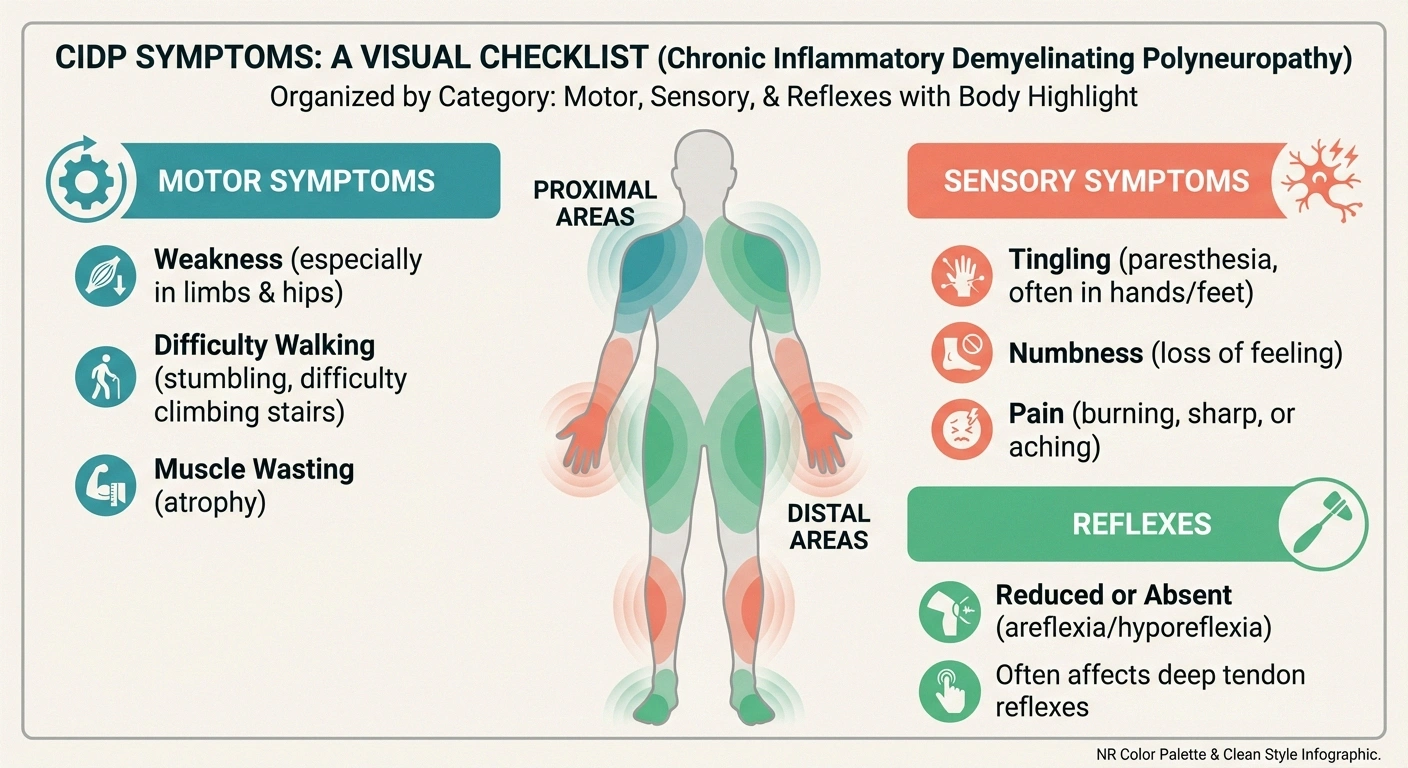
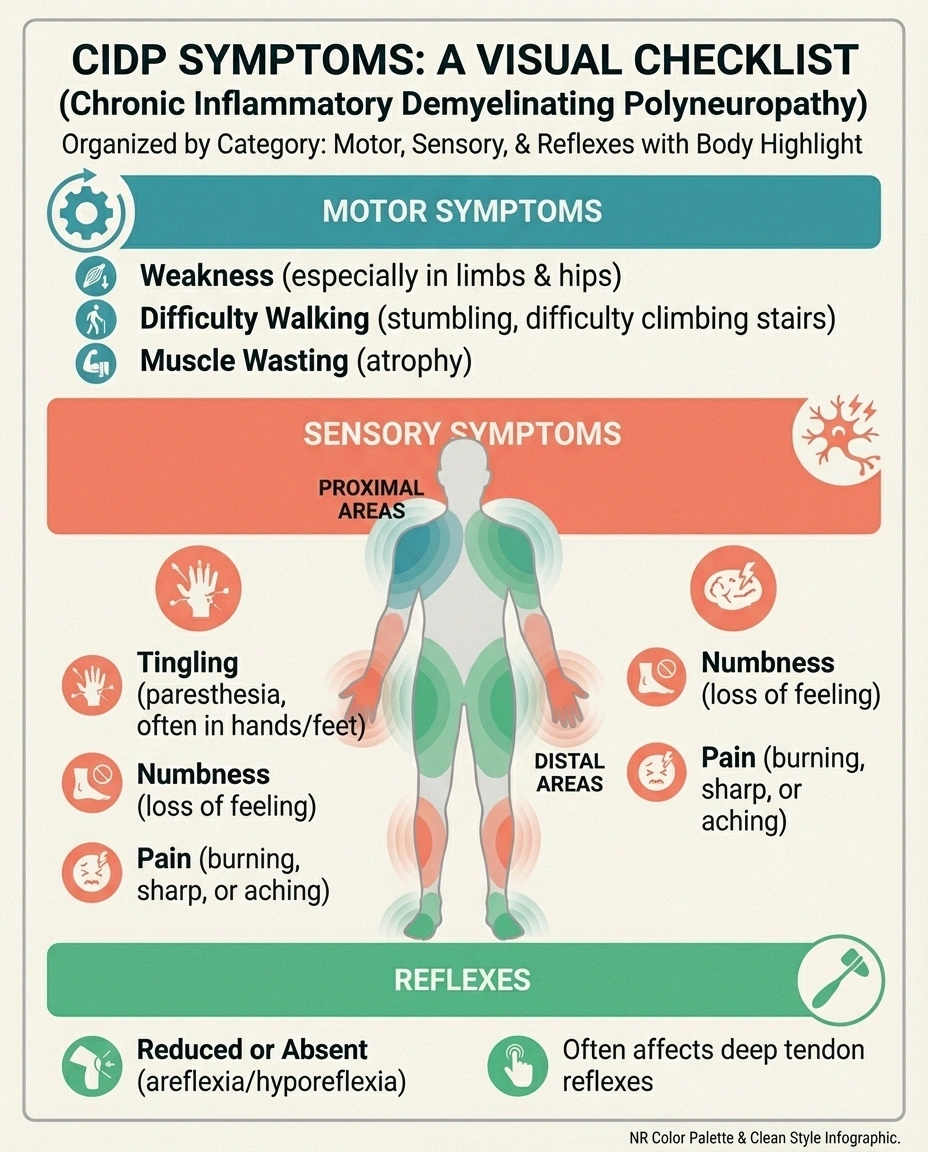
Common symptoms include:
- Progressive weakness in arms and legs, typically on both sides
- Tingling, numbness, or prickling sensations in fingers and toes
- Difficulty walking, climbing stairs, or getting up from a chair
- Loss of balance and coordination
- Reduced or absent reflexes (areflexia)
- Fatigue that goes beyond normal tiredness
- Muscle wasting (atrophy) in affected areas over time
- Neuropathic pain — though this varies widely between individuals
What makes CIDP tricky to recognize is the pace. Because symptoms develop slowly, many people — and their doctors — initially attribute them to aging, deconditioning, or stress. A study published in the Journal of Neurology noted that the average time from symptom onset to CIDP diagnosis can be over a year, with some patients waiting considerably longer.
If you're experiencing unexplained nerve symptoms that keep getting worse over weeks or months — especially if they involve weakness on both sides of your body — bring up CIDP with your doctor. The earlier it's caught, the better the outcome.
CIDP Variants: It Doesn't Always Look the Same
One reason CIDP gets missed is that it doesn't always follow the textbook pattern. Researchers have identified several variants, each with a somewhat different presentation:
Typical CIDP — Symmetrical weakness and sensory changes affecting both arms and legs. This is the most commonly diagnosed form.
Lewis-Sumner Syndrome (Multifocal Acquired Demyelinating Sensory and Motor Neuropathy) — An asymmetric form where weakness and sensory loss affect individual nerves rather than both sides equally. It can mimic conditions like multifocal motor neuropathy.
Pure Sensory CIDP — Numbness, pain, and balance problems without noticeable weakness. People with this variant often struggle with walking and coordination but may have normal-looking muscle strength on examination.
Pure Motor CIDP — Weakness and lost reflexes without numbness or tingling. This variant can be confused with motor neuron disease.
Distal CIDP (DADS) — Distal Acquired Demyelinating Symmetric neuropathy primarily affects the hands and feet, similar to more common peripheral neuropathies. It's sometimes associated with an abnormal blood protein called an IgM paraprotein.
These variants explain why some people with CIDP receive a different diagnosis first — small fiber neuropathy, idiopathic neuropathy, or even fibromyalgia — before the correct condition is identified. If your neuropathy isn't responding to standard treatments, asking about CIDP variants may be worth the conversation.
What Causes CIDP?
The honest answer is that researchers still don't fully understand what triggers CIDP. What they do know is that it's an autoimmune process — your immune system mistakenly identifies the myelin sheath as a threat and attacks it, much like what happens in other autoimmune conditions that damage the nervous system.
Several factors may play a role:
- Immune system dysregulation — Both T cells and antibodies appear to participate in the attack on myelin
- Preceding infection — Some cases follow a viral or bacterial illness, though this connection is less clear than with GBS
- Genetic predisposition — Certain immune system genes may increase susceptibility
- Other autoimmune conditions — CIDP sometimes occurs alongside diabetes, lupus, or inflammatory bowel disease
Unlike diabetic neuropathy where the cause is clearly linked to blood sugar damage, or alcoholic neuropathy where toxic exposure is the culprit, CIDP can appear without any obvious trigger. This uncertainty can be frustrating, but the good news is that treatment doesn't require knowing the exact cause — because we're targeting the immune response itself.
How CIDP Is Diagnosed
Diagnosing CIDP requires putting together several pieces of the puzzle. No single test confirms it — instead, your neurologist will look at the overall picture.
Nerve conduction studies and EMG — These are usually the first diagnostic tests. In CIDP, nerve conduction studies show a specific pattern of demyelination: slowed nerve conduction velocities, prolonged distal latencies, conduction block, and temporal dispersion. This pattern distinguishes CIDP from axonal neuropathies. If you haven't had these tests, our guide on what to expect during EMG and nerve conduction studies can help you prepare.
Lumbar puncture (spinal tap) — The cerebrospinal fluid (CSF) in most CIDP patients shows elevated protein levels with a normal white blood cell count. This finding, called albuminocytologic dissociation, supports the diagnosis.
Blood tests — These help rule out other conditions that can mimic CIDP, including diabetes, vitamin deficiencies, thyroid problems, HIV, hepatitis, and blood disorders. Your doctor may also check for paraproteins — abnormal antibodies that can be associated with certain CIDP variants.
MRI — Magnetic resonance imaging of the nerve roots and brachial plexus may show nerve enlargement or enhancement, which supports the diagnosis. This is particularly useful in atypical cases.
Nerve biopsy — Rarely needed, but in uncertain cases, a nerve biopsy can confirm demyelination and inflammation. This is typically reserved for situations where other tests are inconclusive.
Treatment response — Sometimes the diagnosis is confirmed by how you respond to treatment. If immunotherapy improves your symptoms, it strongly supports a CIDP diagnosis.
The European Academy of Neurology and Peripheral Nerve Society (EAN/PNS) published updated diagnostic criteria in 2021 that many neurologists now follow. These criteria classify cases as “definite,” “probable,” or “possible” CIDP based on clinical and electrodiagnostic findings.
Treatment Options for CIDP
Here's the encouraging part: CIDP is one of the most treatable forms of neuropathy. Because the immune system is driving the damage, treatments that modulate the immune response can often slow, stop, or even reverse the progression.
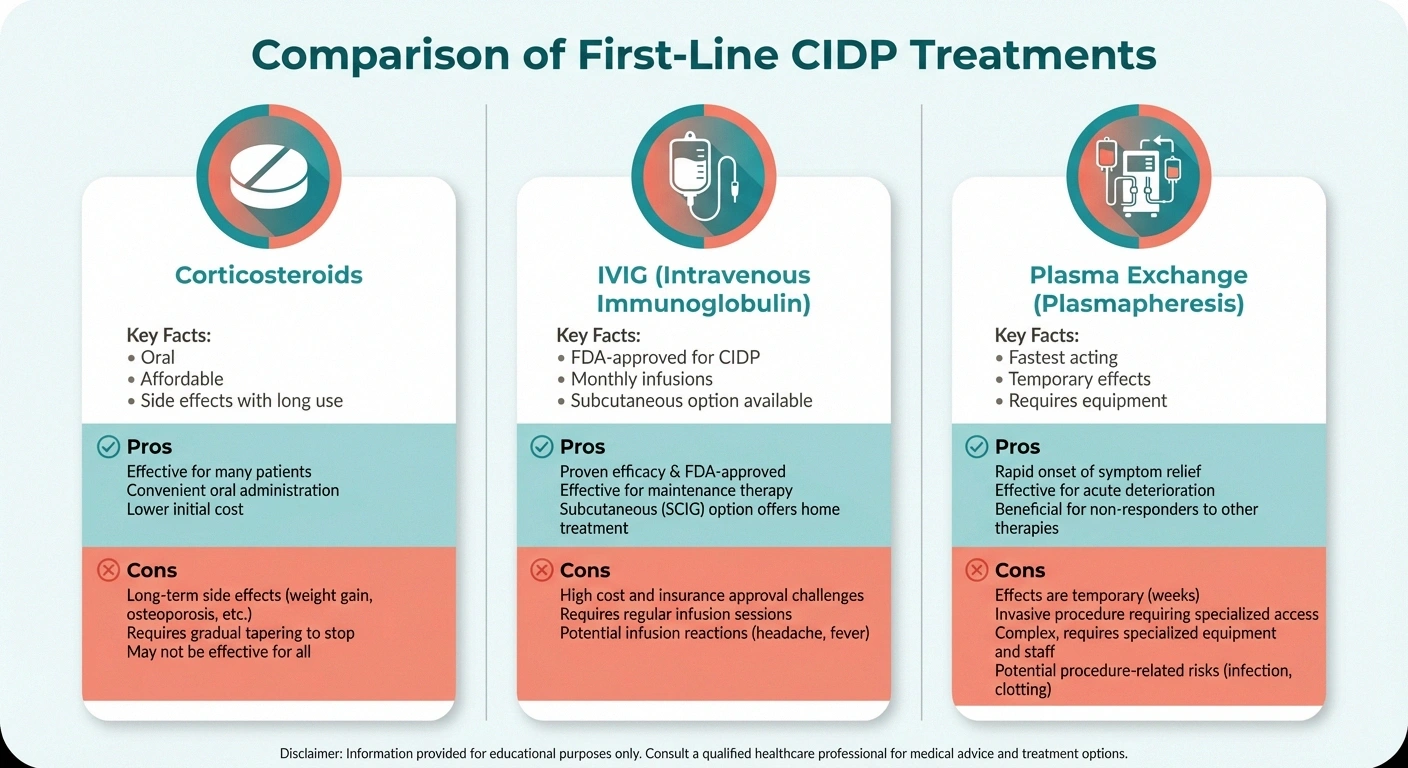

There are three established first-line treatments:
Corticosteroids
Prednisone or prednisolone is often the starting point. These medications suppress the immune response broadly, reducing the attack on myelin. Many patients notice improvement within weeks. The challenge is long-term use — extended courses of steroids carry significant side effects including weight gain, bone thinning, elevated blood sugar, mood changes, and increased infection risk. Most neurologists aim to taper steroids gradually once symptoms improve.
Intravenous Immunoglobulin (IVIG)
IVIG is the only treatment with FDA approval specifically for CIDP. It contains pooled antibodies from healthy donors and works by modulating the immune system in multiple ways. IVIG is typically given as an infusion over several hours, initially as a loading dose followed by maintenance infusions every 3 to 6 weeks. Subcutaneous immunoglobulin (SCIg) is a newer option that some patients can self-administer at home, which offers more flexibility. Side effects are generally milder than steroids but can include headaches, nausea, and rarely, blood clots.
Plasma Exchange (Plasmapheresis)
This procedure filters the blood plasma to remove harmful antibodies. It works quickly — often faster than steroids or IVIG — but the effects are temporary, so it's usually repeated regularly or used as a bridge to other treatments. Plasma exchange requires specialized equipment and venous access, making it less convenient than the other options.
When first-line treatments aren't enough, neurologists may turn to second-line immunosuppressants such as azathioprine, mycophenolate mofetil, or rituximab. These are used when the standard approaches fail, cause intolerable side effects, or don't produce an adequate response.
An important point: CIDP treatment is often described as an art as much as a science. The right combination of therapies, dosing schedules, and maintenance plans varies enormously between individuals. Working with a neurologist who has experience treating CIDP — ideally at a Center of Excellence — can make a meaningful difference in outcomes.
Living with CIDP: What to Expect Long-Term
The long-term outlook for CIDP varies widely. According to data compiled by the GBS-CIDP Foundation, roughly one-third of patients achieve full remission with treatment, one-third have a relapsing-remitting course requiring ongoing therapy, and one-third experience some degree of progressive disability despite treatment.
Several factors influence your prognosis:
- Early diagnosis and treatment — The sooner treatment begins, the less nerve damage accumulates. Prolonged untreated CIDP can cause secondary axonal damage that may be irreversible.
- Treatment response — Patients who respond well to first-line therapies generally have better long-term outcomes.
- CIDP variant — Typical CIDP and Lewis-Sumner syndrome tend to respond better to treatment than distal variants associated with paraproteins.
- Age at onset — Younger patients tend to have better recovery, though CIDP can be successfully treated at any age.
For many people with CIDP, treatment becomes a long-term commitment. This can mean regular IVIG infusions, careful steroid management, or periodic adjustments to immunosuppressive regimens. Building a relationship with a trusted neurologist and establishing a sustainable treatment plan is essential.
Coping Strategies and Practical Tips
Beyond medical treatment, living well with CIDP often involves adjustments to daily life:

Physical therapy — Working with a physical therapist who understands neuromuscular conditions can help maintain strength, improve balance, and prevent falls. Gentle, consistent exercise is usually recommended — pushing through fatigue can worsen symptoms.
Occupational therapy — An occupational therapist can help with hand weakness, adaptive devices, and strategies for maintaining independence with daily activities.
Mental health support — Living with a chronic, fluctuating condition takes a psychological toll. Addressing the emotional side of neuropathy — whether through therapy, support groups, or simply talking to others who understand — is just as important as managing the physical symptoms.
Community connection — The GBS-CIDP Foundation offers support groups, educational resources, and connections to other patients. Hearing from people who've navigated similar challenges can be incredibly reassuring — especially during the early, uncertain phase after diagnosis.
Workplace planning — If CIDP affects your ability to work, explore workplace accommodations under the ADA or disability benefit options early. Having a plan reduces stress during flares.
When to See a Doctor
If you're experiencing progressive weakness or numbness that has been worsening over several weeks — especially if it affects both sides of your body — please don't wait. Early evaluation is critical. Bring up the possibility of CIDP with your neurologist, especially if:
- Standard neuropathy treatments aren't helping
- Weakness is affecting your hips, shoulders, or proximal muscles (not just hands and feet)
- Your reflexes are diminished or absent
- Your symptoms fluctuate — improving and then worsening over time
- You have another autoimmune condition
Ask about nerve conduction studies if you haven't had them. A proper diagnostic workup is the first step toward the right treatment. And if your local neurologist isn't familiar with CIDP, consider seeking a second opinion at a neuromuscular specialist or Center of Excellence.
Frequently Asked Questions About CIDP
Is CIDP the same as Guillain-Barré Syndrome?
No, though they share similarities. Both are immune-mediated demyelinating neuropathies, but GBS develops rapidly over days to weeks and usually resolves, while CIDP develops over at least eight weeks and tends to be chronic. CIDP is sometimes described as the chronic form of GBS, though they are classified as separate conditions.
Can CIDP be cured?
Some patients achieve full remission and can eventually stop treatment. However, many require ongoing therapy to keep symptoms controlled. Early treatment gives you the best chance of a favorable outcome, which is why prompt diagnosis matters so much.
How long does CIDP treatment take to work?
Response times vary by treatment. Corticosteroids and IVIG may show improvement within weeks, while plasma exchange often works within days. Some patients notice gradual improvement over months. If one treatment isn't working after an adequate trial, your neurologist may try a different approach.
Does CIDP affect life expectancy?
CIDP itself is not typically life-threatening with appropriate treatment. The primary concern is disability from progressive weakness if the condition goes untreated or doesn't respond to therapy. With proper management, most people with CIDP live normal lifespans.
Can diet or supplements help CIDP?
While no specific diet has been proven to treat CIDP, maintaining good nutrition supports overall nerve health and immune function. Some patients find that an anti-inflammatory diet helps manage symptoms alongside medical treatment. Supplements should be discussed with your neurologist, as some may interact with immunosuppressive medications.
Is CIDP hereditary?
CIDP is not directly inherited, but certain genetic factors may make some people more susceptible to autoimmune conditions in general. If autoimmune diseases run in your family, mention this to your doctor — it can be a useful clue during the diagnostic process.
The Bottom Line
CIDP is a rare but treatable autoimmune neuropathy that deserves to be on more people's radar — patients and doctors alike. The biggest enemy isn't the disease itself but delayed diagnosis. When treatment starts early, many people with CIDP maintain excellent quality of life. If you suspect CIDP or have been recently diagnosed, find a neurologist with neuromuscular expertise, connect with the GBS-CIDP Foundation for support, and remember that you're not navigating this alone.
For more about how neuropathy progresses and what different stages mean, visit our comprehensive guide on the stages of neuropathy. And if you're looking for practical advice on living with chronic nerve conditions, browse our living with neuropathy resources.


